






 本文介绍了蛋白质语言模型在预测蛋白质结构和功能方面的应用,包括基于Transformer的ESM-1b、ESM-MSA-1b和ESM-1v模型。这些模型通过无监督学习直接从氨基酸序列中学习结构信息,进行结构预测、功能预测和序列变异影响评估。ESM-1v模型实现了zero-shot预测,无需针对特定家族额外训练,提高了预测效率和准确性。
本文介绍了蛋白质语言模型在预测蛋白质结构和功能方面的应用,包括基于Transformer的ESM-1b、ESM-MSA-1b和ESM-1v模型。这些模型通过无监督学习直接从氨基酸序列中学习结构信息,进行结构预测、功能预测和序列变异影响评估。ESM-1v模型实现了zero-shot预测,无需针对特定家族额外训练,提高了预测效率和准确性。
一、背景
现阶段蛋白序列数据库正呈指数级增长,而目前人们已经了解其结构的蛋白质仅占数据库的一小部分。若能够仅通过蛋白质的氨基酸序列预测蛋白质结构与功能,将大大提高生化研究的效率,并有助于蛋白质设计技术的发展。
语言模型(language model)是能够基于之前的文本预测下一个字符或词汇的一类神经网络,能够学习某种语言中各字符、词汇的统计规律并生成符合相应规律的新序列。语言模型涵盖部分循环、卷积神经网络以及基于Transformer的各类模型,广泛应用于自然语言处理领域。蛋白质语言模型(protein language model)是各类语言模型在生物化学领域的迁移应用,输入蛋白序列并学习序列中隐含的生化性质、二三级结构、功能内在规律,能够完成预测蛋白结构、预测蛋白功能、生成新序列等任务。
通过语言模型预测蛋白结构的原理:生物体的蛋白序列不是氨基酸的随机排列组合,而是受到自然选择的制约,即长期的自然选择决定了蛋白序列具有某种统计规律。一方面,蛋白序列在其家族中的变异规律能反映其结构,若某位点在进化过程中保守地选择某一种或某几种氨基酸,说明只有这几种氨基酸的生化性质能适应该蛋白在此处的结构;另一方面,在一条序列中,若某些位点不是独立进化而是相互制约,说明它们很可能在蛋白的三级折叠结构中相互作用。也就是说,蛋白序列的统计规律隐含其结构信息,而语言模型恰能通过输入序列学习这种规律,因此,通过语言模型能够预测蛋白质的结构。
蛋白质序列处理与自然语言处理问题的重要区别是,蛋白序列主要由20中氨基酸构成,而不像自然语言那样有丰富的词汇。蛋白序列中的氨基酸可当做字符进行处理,因此蛋白质序列处理可看做字符级的自然语言处理问题。
二、内容概述
ESM系列三篇文章的主线是利用蛋白质语言模型实现从蛋白序列预测蛋白质结构和功能,提出了ESM-1b、ESM-MSA-1b、ESM-1v三种基于Transformer的无监督的蛋白质语言模型。无监督的语言模型能够直接利用蛋白数据库中的海量序列信息,而不依赖对序列的人工注释。
第一篇《Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences 》介绍了该团队基于Transformer训练的顶尖水准(state-of-the-art)蛋白质语言模型ESM-1b,能够直接通过蛋白的氨基酸序列预测该蛋白的结构、功能等性质。
第二篇《MSA Transformer》在ESM-1b的基础上作出改进,将模型的输入从单一蛋白质序列改为MSA矩阵,并在Transformer中加入行、列两种轴向注意力机制,对位点分别计算第
个序列和第
个对齐位置的影响,充分利用二维输入的优势。
第三篇《Language models enable zero-shot prediction of the effects of mutations on protein function 》中提出了ESM-1v模型,该模型与ESM-1b模型构架相同,只是预训练数据集改为UR90(ESM-1b预训练数据集为UR50)。ESM-1v为一种通用的蛋白质语言模型,能够实现蛋白质功能的zero-shot预测,即模型只需经过预训练即可应用于各种具体问题,对于特定蛋白质预测问题(例如针对特定蛋白家族)无需额外训练即可直接解决。

三个模型的对比:
| 模型名称 | 输入数据类型 | 普适性 |
|---|---|---|
| ESM-1b | single sequence | family-specific |
| ESM-MSA-1b | MSA | few-shot |
| ESM-1v | single sequence | zero-shot |
三、内容梳理
(一)ESM-1b模型
1. 模型描述
ESM-1b实际上是以蛋白质序列作为输入,经过超参数(hyperparameter)优化,训练的一个高模型容量(high-capacity)的Transformer。经过训练后,该模型输出的特征表示(representation)中隐含蛋白质的二三级结构、功能、同源性等信息,并且这些信息能够通过线性投影(linear projection)显化。
Transformer模型适应蛋白质预测问题、能够作为蛋白质语言模型基础的原因猜想:Transformer的self-attention机制能够直接计算序列中残基之间的两两联系(pair-wise interaction),能够捕捉不同位置的氨基酸残基之间的相互依赖和作用关系,这种作用关系恰恰是由蛋白质的结构决定的,是蛋白结构在序列模式中的反映。
2. 模型结构
ESM-1b模型实际上是参数经过调整的Transformer,采用系统优化法优化Transformer中的超参数,再在UR50数据库上进行预训练,即可得ESM-1b模型。
3. 模型训练
(1)训练方法
本模型的最终目标是探究Transformer能否从蛋白序列中提取结构信息,但该目标不容易直接通过训练实现,因此模型训练使用的是代理任务——masked training,即随机遮盖序列中部分片段,基于序列中其他未被遮盖的残基预测被遮盖部分真实的残基是什么。
这种代理训练任务能够将蛋白结构特性嵌入特征表示的原因猜想:
要根据序列中其他处七年级预测masked token的实际值,就要学习序列中残基之间的联系,而残基联系是蛋白结构在序列中的反映,因此学习了残基联系也就学习了结构功能。
(2)训练数据:UR50/S
(3)训练参数
- 参数规模:650M
- 层数:33
3. 训练结果解读
经过训练,在ESM-1b模型的特征表示中能够解读出多维度的蛋白质相关信息,可以通过对比模型训练前后的特征表示来解读这些信息。
(1)残基生化特性
模型的输出嵌入(output embedding)可以看作n维向量,而n维向量又可以看作n维空间中的散点。将这些点通过t-SNE方法降维到二维,发现具有相似生化特征的氨基酸残基在二维平面坐标中被聚成一类(如疏水的、极性的、芳香的、正电性的、负电性的等)。而未训练的特征表示中不具有这样的特性。
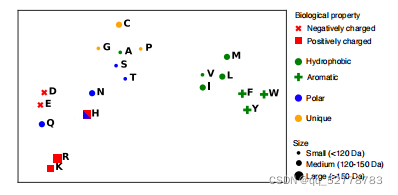
(2)序列变异特性
与(1)相同,将每个蛋白看作n维空间的点,同样将这些点通过t-SNE方法降维到二维,发现代表直系同源基因的蛋白被聚成一类。在二维空间中,沿横轴蛋白被聚成不同物种,沿纵轴蛋白被聚成不同直系同源基因。而未训练的特征表示中蛋白散乱分布。

(3)远缘同源性
基于训练后的特征表示,用SCOPe方法预测蛋白是否属于同一superfamily或fold,预测结果全部正确。
(4)二级结构
基于ESM-1b的特征表示,以蛋白的8种二级结构作为标签,拟合logistic回归模型来预测蛋白序列的二级结构。
(5)三级结构
为序列两个位点分别做线性投影,二者做点积,得到一个二元变量,1表示两位点在蛋白质三级结构中有联系,0表示二者在三级结构中无联系。
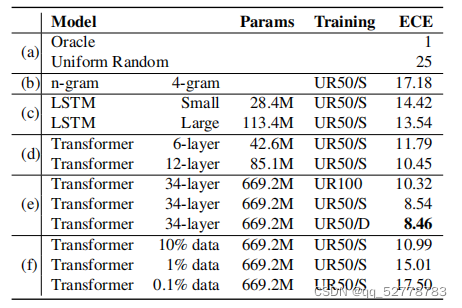
4. 模型评估
以交叉熵(ECE)作为标准,比较不同语言模型的训练效果,交叉熵越小则效果越好。本文比较了n-gram、LSTM和Transformer三种模型的表现,发现仅仅6层的Transformer模型即可达到比LSTM模型更优的效果;进一步探究发现Transformer,层数越大交叉熵越小,训练数据丰富度越大交叉熵越小。在数据集UR50/D上,34层Transformer交叉熵可降低至8.46,达到了SOTA(state-of-the-art)级别。
(二)ESM-MSA-1b模型(MSA Transformer)
MSA Transformer基于一个蛋白家族的MSA矩阵,训练针对该家族的具体模型,训练之后能够在Transformer的内部结构中(attention map)找到有关蛋白结构的信息,并基于此进行预测。
1. 模型输入
MSA Transformer的输入为一系列二维MSA矩阵,每个MSA是一组蛋白序列的多序列比对文件。
MSA矩阵存储来自不同物种的同类蛋白序列。矩阵的每一行是一条来自特定物种的蛋白序列;每一列在该类蛋白中处于同一位置,记录不同物种在该位置的氨基酸取值。
(1)词嵌入
将不同氨基酸用整数表示,形成一个整型向量。词库包括20中标准氨基酸、5种非标准氨基酸和4种特殊字符,共29个数字。
(2)位置嵌入
- sequence embedding
为序列的每个位置,也就是每一列赋一个序号。
- position embedding
为每条序列,也就是每一行赋一个序号。
位置嵌入使得MSA中的行和列有独一无二的序号,每一行或每一列将被识别为不同的。
2. 模型输出
模型使用masked training方法进行训练,直接输出的是每一个masked token处为各种氨基酸的概率。然而本文的主要目标并不是得到这些概率,而是要通过训练后的attention map预测蛋白质二、三级结构。
(1)二级结构预测(second structure prediction)
训练一个Netsurf模型,基于MSA Transformer的特征表示向量(representation)预测蛋白质的8种蛋白结构,准确率为72.9%。
(2)三级结构预测(contact prediction)
基于MSA Transformer各层、各注意力头的attention map,训练logistic回归模型,对蛋白质三级结构进行预测。
本文使用的Transformer共12层,每层12个注意力头,在总共的144个注意力头中,有55个能够有效预测蛋白质的三级结构(主要位于最后一层,45个行注意力头)。
3. 模型结构
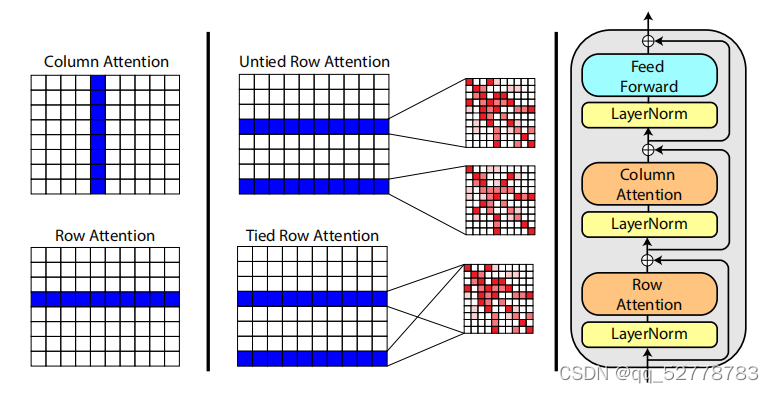
单层MSA Transformer的结构如上图所示,与原始Transformer的结构类似,由注意力与前馈神经网络两部分组成。
(1)轴向注意力机制(axial attention)
针对二维输入数据的特征,本文提出两种轴向注意力机制:行注意力(row attention)和列注意力(column attention)。为了降低算法的时间复杂度,轴向注意力将来自二维平面的注意力分解为纵、横两个方向。轴向注意力认为MSA中某位点受到其他位点的影响主要来自于其所在的行和列,而矩阵中其他位置的影响可以忽略不计,对位点只需计算第
行和第
列对它的attention权重。
行注意力计算位点所在行的注意力权重,本质是提取该点所在蛋白序列的信息,计算同一蛋白序列中不同氨基酸之间的相互影响权值,能够反应这些氨基酸在蛋白二、三级结构中的相互作用,进而推测蛋白结构。
列注意力计算位点所在列的注意力权重,本质是提取该位置上氨基酸的共变信息。通过不同物种序列在相同位置氨基酸选择的直观对比,能够用更少的参数解读该位置氨基酸在整个家族中的变异和进化规律。(对于一单一蛋白序列为输入的语言模型,虽然也能从单一序列中解读氨基酸进化规律,但需要更多的参数支持。)
行注意力计算公式:
其中表示一个MSA中的序列条数,除以
的方法称为平方根标准化(square-root normalization),其中除以
是为了消除序列条数对注意力权重大小的影响,除以
与原始Transformer中目的相同:获得更稳定的梯度。
(2)前馈神经网络
在计算横向、纵向注意力之后,设置一层前馈神经网络(更接近Transformer解码器的结构)。
4. 模型训练
(1)训练方法
使用masked language modeling方法进行训练,将蛋白的部分序列片段遮盖或替换成特定token,进行自监督的masked training。
损失计算公式:
(2)训练数据
训练数据库为UR50,数据量达4.3TB,含有26million MSA,平均每个MSA含有1192条蛋白序列。
(3)训练参数
- 参数规模:100M(性能超过650M参数的Potts模型和蛋白质语言模型)
- 层数:12
- embedding size:768
- attention head数:12
- batch size:512(一个batch含512个MSA)
- 学习率:
5. 模型性能分析
(1)与其他模型对比的表现
氨基酸的接触模式(contact pattern)能够直接被行注意力头学习,蕴含在行注意力的attention map中。
基于训练发现的接触模式,MSA Transformer能够对蛋白质的接触情况进行预测(contact prediction),预测准确率显著优于Potts等其他模型(不论输入MSA的深度如何),尤其是在MSA深度较小时,优势更加明显。
(2)探究MSA Transformer预测蛋白结构的机制
①学习协方差(与Potts模型一致)
随机打乱MSA每一列氨基酸的排列,破坏该列的协方差特性,发现Potts模型预测准确率降到随机预测(random guess)级别,ESM-1b预测结果不受影响,MSA Tansformer预测准确率下降,但仍有有效预测能力。说明MSA Transformer能够学习协方差特性,但不唯一依赖于协方差特性。
②学习序列规律(与ESM-1b一致)
随机打乱MSA中列的排列顺序而保持每一列内部不变,破坏每条序列的规律,发现ESM-1b模型预测准确率降到随机预测(random guess)级别,Potts模型预测结果不受影响,MSA Tansformer预测准确率下降,但仍有有效预测能力。说明MSA Transformer能够学习序列规律,但不唯一依赖于序列规律。
综上,MSA Transformer能够同时学习MSA中每列的协方差特性和每一行的序列规律,能够充分利用MSA中的信息。
(三)ESM-1v模型
1. 模型描述
ESM-1v是在数据集UR90上训练的通用蛋白质语言模型,相对之前模型的突破点在于能够对序列变异对蛋白质功能的影响进行zero-shot预测。
zero-shot预测指的是,一个预测模型只需要针对某个特定问题做预训练,不需要其他的额外训练就可以直接迁移到其他问题。例如某个翻译模型在执行将A语言翻译成B语言的训练后,无需任何额外学习,即可完成语言A到语言C的翻译。
在ESM-1v模型之前,虽然蛋白质语言模型是采用无监督方法训练的,但在执行下游任务时也需要用实验数据进行监督。比如要基于蛋白质语言模型的特征预测蛋白质结构,需要基于已知的蛋白质结构进行监督训练,如逻辑回归;即使是MSA Transformer,也收到一小部分来自MSA的监督,因为MSA相当于提供了一系列可行的变异范本(positve examples)。
zero-shot要求模型在预训练阶段就学习到之后的具体任务所需的一切信息。本文中使蛋白质语言模型具有zero-shot预测能力的机制是,采用含有海量进化信息的蛋白数据库进行预训练。当所用数据库涵盖的序列足够多、足够多样(large and diverse),那么模型就有可能从数据库中学到横跨整个进化树的序列模式,那么该模型也就很可能会在预训练阶段学习到它将要应用的家族的序列模式,迁移应用时也就无需再额外训练。
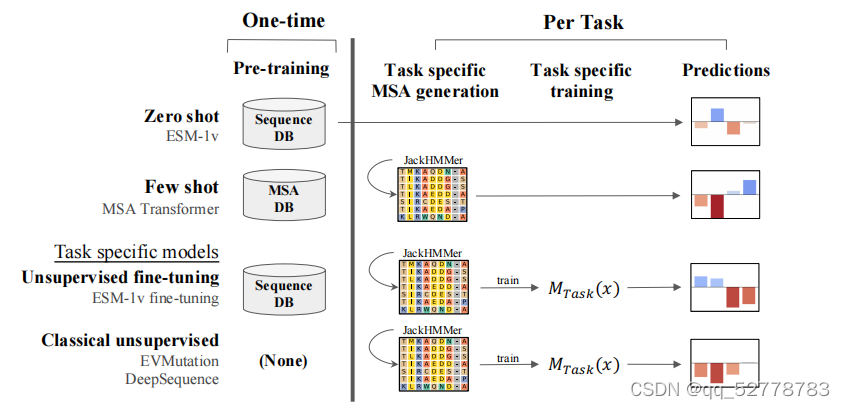
ESM-1v不需要生成某个家族的MSA,连MSA的监督都不需要
2. 模型原理
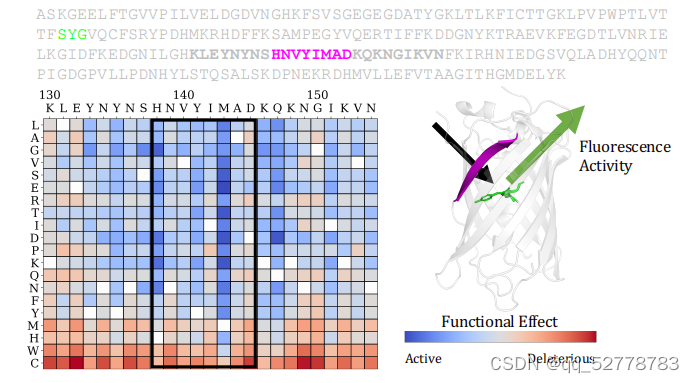
用氨基酸的保守性衡量变异的影响力,若某位置与野生型相比其他氨基酸出现的概率很低,说明蛋白序列在该位点很保守,变异可能性很低,也就说明该位点的氨基酸可能对蛋白的结构和功能有重要影响。
预测序列变异对蛋白功能的影响的方法:为每个变异对功能的影响打分来衡量其影响,比较某个被遮盖位置(masked)变异成某个氨基酸的概率和该位置变异成野生型蛋白的概率,求它们的log-odds,即求它们概率的比值再取对数。
打分公式如下:
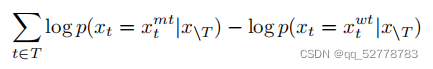
3. 模型结构
ESM-1v模型采用与ESM-1b相同的结构。
4. 模型训练
(1)训练方法:masked training
(2)训练数据
UR90数据库,含有98million条多样的蛋白序列,数据量和数据多样性都远远大于ESM-1b和MSA Transformer训练所用的UR50数据库,因此训练出的模型迁移能力更强。
(3)训练参数规模:650M
4. 模型评估
(1)准确率
用模型预测结果与实验室测量结果的相关性衡量模型预测的准确率,这种相关性用spearman相关系数表示。ESM-1v的表现在17个数据集上由于传统的DeepSequence模型,在zero-shot任务中的表现优于ProtBERT-BFD、ESM-1b等蛋白质语言模型。
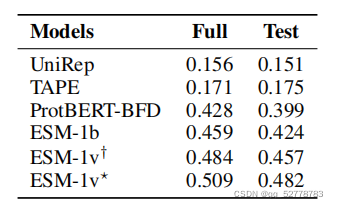
(2)执行效率
执行效率高与其他蛋白质语言模型,原因有二:其一,ESM-1v模型在迁移应用时无需额外训练,节省了时间;其二,ESM-1v模型在训练过程中只有前向传播(forward pass)过程而没有梯度更新(gradient update)过程(猜想是由于缺少监督所以无法更新梯度)。

















 9659
9659

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









