






VCF文件包含了基因组中的变异信息以及与样本相关的数据。对于BSA(Bulked Segregant Analysis)重测序分析,我们希望更直观地查看各个样本在特定位点的变异信息,包括变异深度和变异频率。为了实现这一目标,本次编写了一个Python脚本,用于提取和重新组织VCF文件中的数据。
这个脚本的主要功能包括:
1)从VCF文件中解析数据,包括染色体位置、参考碱基、可变碱基等。
2)提取每个样本的基因型信息,包括杂合、纯合等。
3)获取每个样本的变异深度和变异频率。
4)动态构建基因型映射,以确定每个样本的变异类型。
5)将处理后的数据按特定格式重新组织。
6)将处理后的数据写入输出文件,以便进一步分析和可视化。
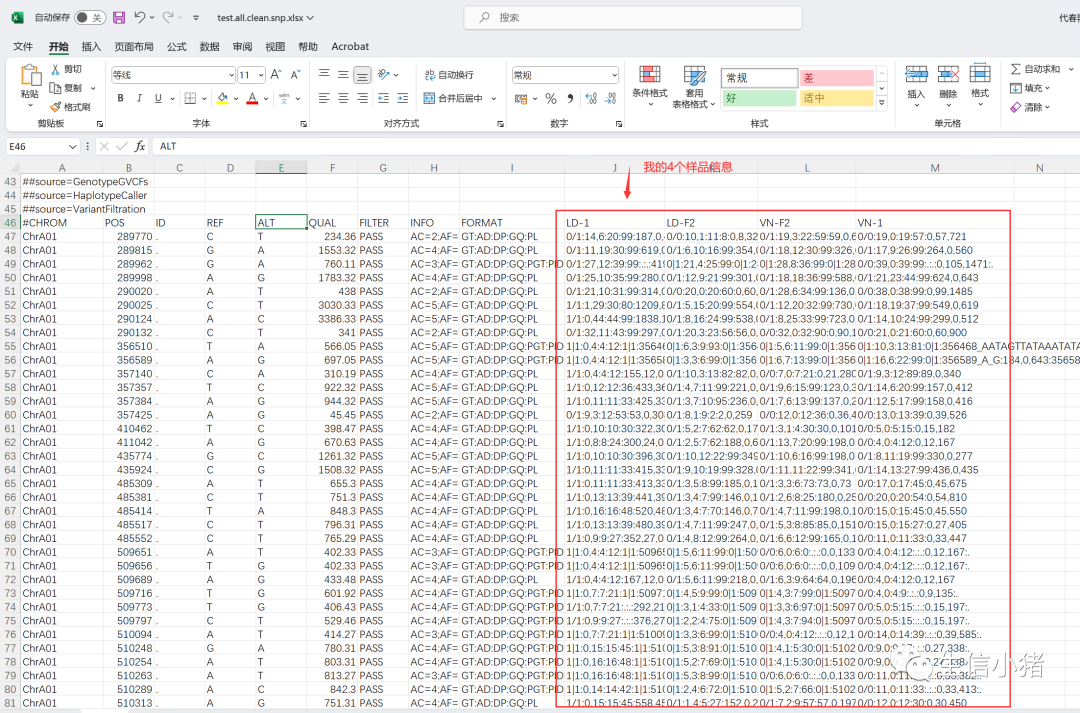
下面是我的vcf文件内容,因为在vcf文件里看起来不太方便,这里我把vcf文件里的内容复制到excel中看一下。
下面是经过脚本处理后输出的文件
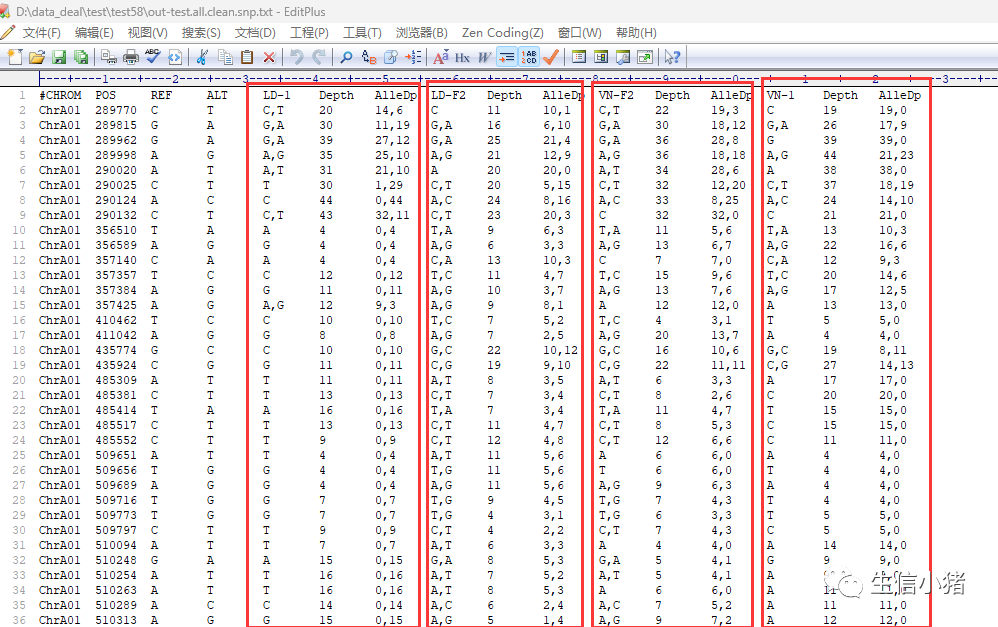
这样我们就可以以这种方式去看各个样本相对于参考基因组的变异以及样本之间的变异了。
一、在windows下rstudio中的运行脚本
input_file = 'D:/data_deal/test/test58/test.all.clean.snp.vcf'
output_file = 'D:/data_deal/test/test58/out-test.all.clean.snp.txt'
# 初始化一个空字典来映射基因型
genotype_mapping = {}
# 初始化一个列表来保存样本名称
sample_names = []
# 初始化一个列表来保存处理后的数据行
output_data = []
with open(input_file, 'r') as infile:
for line in infile:
if line.startswith('#CHROM'):
parts = line.strip().split('\t')
sample_names = parts[9:] # 获取样本名称
# 构建标题行
header = ['#CHROM', 'POS', 'REF', 'ALT']
for sample_name in sample_names:
header.extend([sample_name, 'Depth', 'AlleDp'])
output_data.append(header)
if not line.startswith('#'):
# 分割行,获取所需的列
parts = line.strip().split('\t')
chrom = parts[0]
pos = parts[1]
ref = parts[3]
alt = parts[4]
# 获取样本数据列
sample_data = parts[9:]
# 初始化列表来保存处理后的数据
new_sample_data = []
for data in sample_data:
# 分割样本数据列
sample_parts = data.split(':')
# 提取基因型信息
genotype = sample_parts[0]
# 获取Depth和AlleDp
ad = sample_parts[1].split(',')
depth = sample_parts[2]
alle_dp = ','.join(ad)
# 动态构建基因型映射
genotype_mapping[genotype] = f'{ref},{alt}'
# 处理0/0(0|0)和1/1(1|1)的情况,只写入和REF或ALT列一致的值
if genotype in ('0/0', '0|0'):
base_type = ref
elif genotype in ('1/1', '1|1'):
base_type = alt
else:
base_type = genotype_mapping.get(genotype, '')
# 构建处理后的样本数据列
new_sample_info = [base_type, depth, alle_dp]
new_sample_data.extend(new_sample_info)
# 构建数据行
data_row = [chrom, pos, ref, alt] + new_sample_data
output_data.append(data_row)
# 打开输出文件并将处理后的数据写入
with open(output_file, 'w') as outfile:
for data_row in output_data:
outfile.write('\t'.join(data_row) + '\n')
二、在linux中运行
下面是在linux下具体运行vcf_processor.py步骤
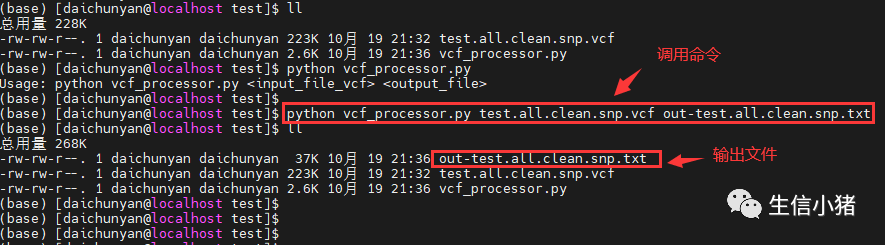
下面是vcf_processor.py脚本的全部内容
import sys
if len(sys.argv) != 3:
print(f"Usage: python {sys.argv[0]} <input_file_vcf> <output_file>")
sys.exit(1)
input_file = sys.argv[1]
output_file = sys.argv[2]
# 初始化一个空字典来映射基因型
genotype_mapping = {}
# 初始化一个列表来保存样本名称
sample_names = []
# 初始化一个列表来保存处理后的数据行
output_data = []
with open(input_file, 'r') as infile:
for line in infile:
if line.startswith('#CHROM'):
parts = line.strip().split('\t')
sample_names = parts[9:] # 获取样本名称
# 构建标题行
header = ['#CHROM', 'POS', 'REF', 'ALT']
for sample_name in sample_names:
header.extend([sample_name, 'Depth', 'AlleDp'])
output_data.append(header)
if not line.startswith('#'):
# 分割行,获取所需的列
parts = line.strip().split('\t')
chrom = parts[0]
pos = parts[1]
ref = parts[3]
alt = parts[4]
# 获取样本数据列
sample_data = parts[9:]
# 初始化列表来保存处理后的数据
new_sample_data = []
for data in sample_data:
# 分割样本数据列
sample_parts = data.split(':')
# 提取基因型信息
genotype = sample_parts[0]
# 获取Depth和AlleDp
ad = sample_parts[1].split(',')
depth = sample_parts[2]
alle_dp = ','.join(ad)
# 动态构建基因型映射
genotype_mapping[genotype] = f'{ref},{alt}'
# 处理0/0(0|0)和1/1(1|1)的情况,只写入和REF或ALT列一致的值
if genotype in ('0/0', '0|0'):
base_type = ref
elif genotype in ('1/1', '1|1'):
base_type = alt
else:
base_type = genotype_mapping.get(genotype, '')
# 构建处理后的样本数据列
new_sample_info = [base_type, depth, alle_dp]
new_sample_data.extend(new_sample_info)
# 构建数据行
data_row = [chrom, pos, ref, alt] + new_sample_data
output_data.append(data_row)
# 打开输出文件并将处理后的数据写入
with open(output_file, 'w') as outfile:
for data_row in output_data:
outfile.write('\t'.join(data_row) + '\n')
整个脚本的思路是从输入的VCF文件的第10列开始处理数据。它首先解析VCF文件中的各列,然后针对每个样本的数据,提取和处理基因型信息、深度信息等,最后将处理后的数据重新组织并写入到输出文件中。
注:本次脚本没有考虑ALT有两个的情况。因为我在前面过滤vcf文件时,使用参数min_alleles 2,max_alleles 2,只保留了vcf文件ALT列只有一种碱基的情况。
补充:FORMAT 为后面10列信息的说明列,通常以":"隔开各个缩写词。不同的变异检测软件可能会有差异,以下用GATK的检测结果为例:
第10列(包含)以后为样品基因型列,各信息以":"分隔与FORMAT列一一对应;
GT 表示genotype,通常用/ or |分隔两个数字,|phase过也就是杂合的两个等位基因知道哪个等位基因来自哪条染色体;0代表参考基因组的碱基类型;1代表ALT碱基类型的第一个碱基(多个碱基用","分隔),2代表ALT第二个碱基。,以此类推;比如 REF列为:A, ALT列为G,T;那么0/1基因型为AG 杂合,1/1基因型为GG纯合SNP;1/2代表GT基因型;./.表示缺失;
AD 两种碱基各自支持的碱基数量,用","分开两个数据,分别代表两个等位基因的深度;
DP 该样品该变异位点的测序深度总和,也就是AD两个数字的和;
PL 归一化后各基因型的可能性,通常有三个数字用','隔开,顺序对应AA,AB,BB基因型,A代表REF,B代表ALT(也就是0/0, 0/1, and 1/1),由于是归一化之后,数值越小代表基因型越可靠;那么最小的数字对应的基因型判读为该样品的最可能的基因型;(provieds the likelihoods of the given genotypes)
GQ针对PL的判读得到的基因型的质量值,此值越大基因型质量值越好。由于PL归一化之后通常最小的数字为0;那么基因型的质量值取PL中第二小的数字,如果第二小的数字大于99,我们只取99,因为在GATK中再大的值是没有意义的,第二小的数大于99的话一般说明基因型的判读是很可靠的,只有当第二小的数小于99的时候,才有必要怀疑基因型的可靠性。
补充内容来自:https://www.omicsclass.com/article/6
之前的脚本忘了考虑vcf里面对0/0和1/1格式的另一种书写形式0|0和1|1了。















 3983
3983











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









