






annovar软件是由perl语言编写的,从官网直接下载后就可直接使用。但是该软件需要学术机构邮箱注册申请下载,申请通过后会收到包含下载链接的邮件。
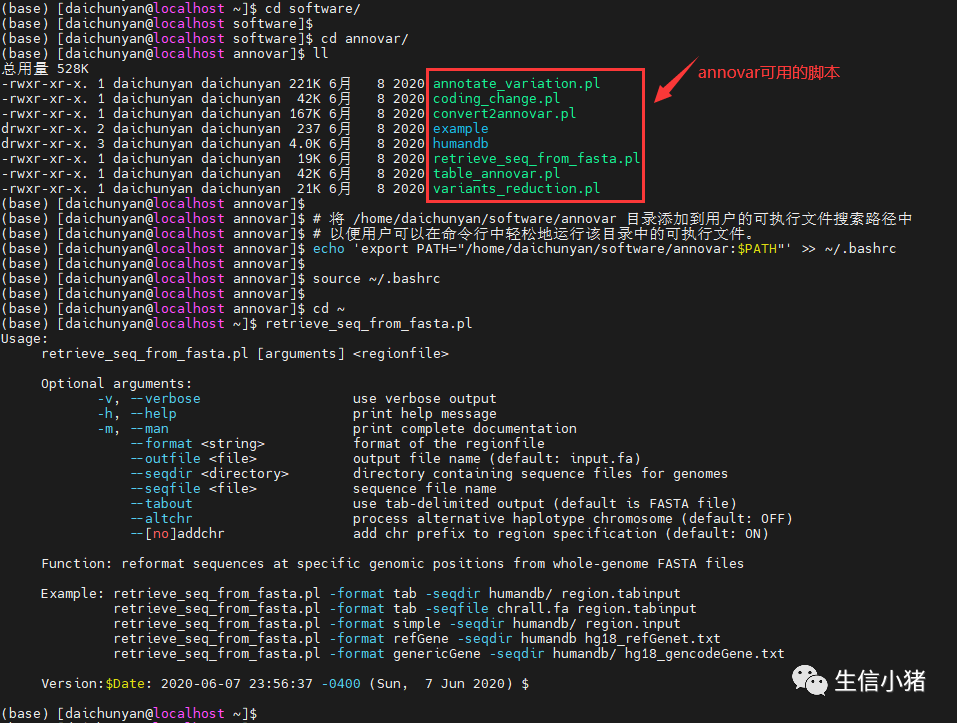

主要包含三种不同的注释方法:gene-based, region-based 和filter-based。
基于基因的注释(Gene-based Annotation)揭示variant与已知基因直接的关系以及对其产生的功能性影响;
基于区域的注释(Region-based Annotation)揭示variant 与不同基因组特定段的关系,例如:它是否落在转录因子结合区域等;
基于筛选的注释(Filter-based Annotation)则分析变异位点是否位于指定的数据库中,比如dbSNP, 1000G,ESP 6500等数据库,计算SIFT/PolyPhen/LRT/MutationTaster/MutationAssessor等。
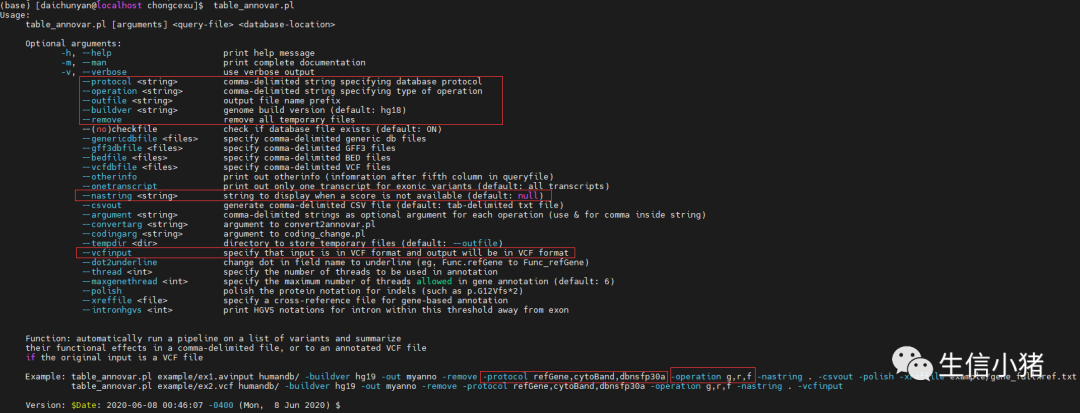
annovar提供了两个脚本以供注释使用:annotate_variation.pl一次注释一个数据库,table_annovar.pl一次注释多个数据库。
一、对基因组文件构建数据库
需要.fa基因组和gff3两个文件
# 转gff3为gtf格式gffread Arabidopsis_thaliana.TAIR10.47.chromosome.4.gff3 -T -o Arabidopsis_thaliana.TAIR10.47.chromosome.4.gtf# 转gtf成refGene格式gtfToGenePred -genePredExt Arabidopsis_thaliana.TAIR10.47.chromosome.4.gtf Arabidopsis_thaliana.TAIR10.47.chromosome.4_refGene.txt# 提取序列文件retrieve_seq_from_fasta.pl --format refGene --seqfile Arabidopsis_thaliana.TAIR10.dna.chromosome.4.fa -outfile Arabidopsis_thaliana.TAIR10.47.chromosome.4_refGeneMrna.fa Arabidopsis_thaliana.TAIR10.47.chromosome.4_refGene.txt
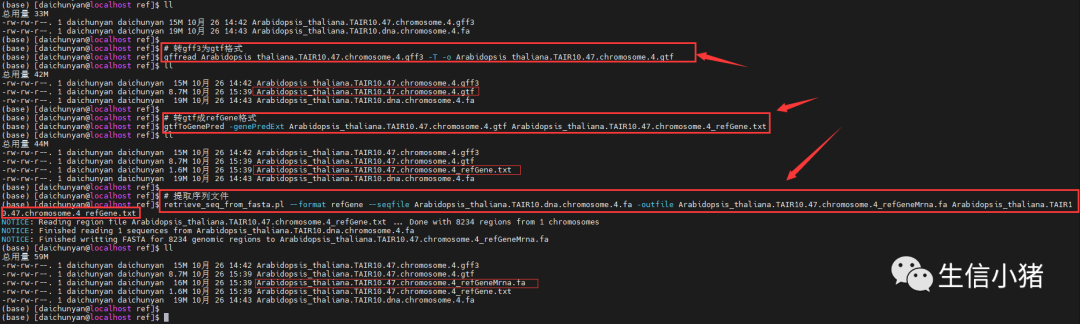
最后输出两个文件Arabidopsis_thaliana.TAIR10.47.chromosome.4_refGeneMrna.fa和Arabidopsis_thaliana.TAIR10.47.chromosome.4_refGene.txt,这两个文件是后续使用annovar注释vcf文件需要用到的文件,记住它的所在路径。
使用annovar提供了两个脚本以供注释使用:annotate_variation.pl一次注释一个数据库,table_annovar.pl一次注释多个数据库。
二、使用table_annovar.pl 一次注释多个数据库
2.1 注释SNP的vcf文件
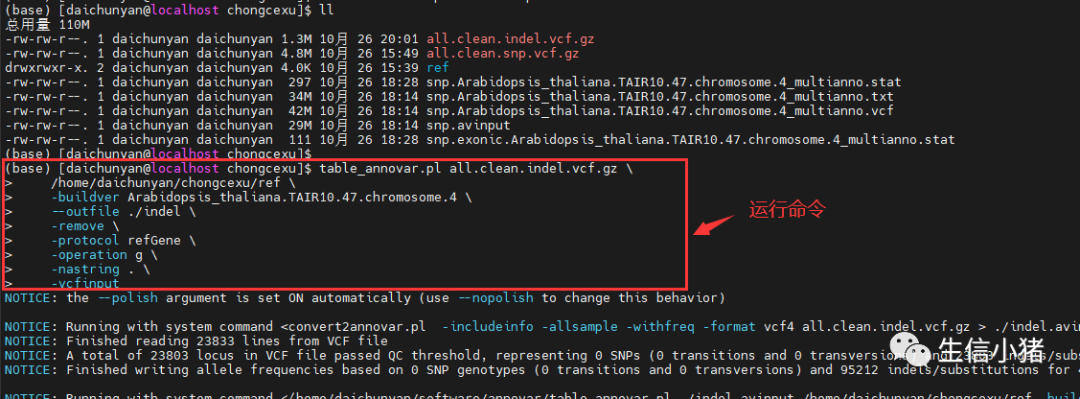
table_annovar.pl all.clean.snp.vcf.gz \/home/daichunyan/chongcexu/ref \-buildver Arabidopsis_thaliana.TAIR10.47.chromosome.4 \--outfile ./snp \-remove \-protocol refGene \-operation g \-nastring . \-vcfinput
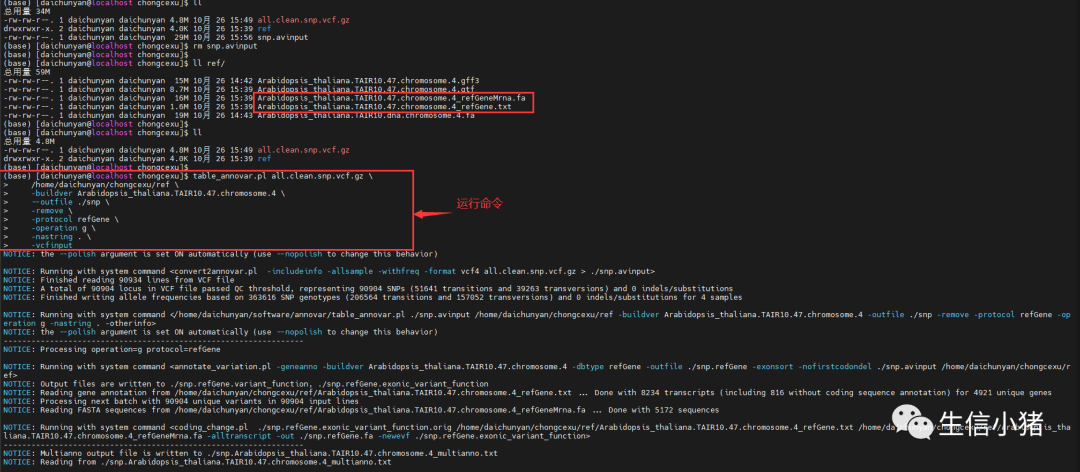
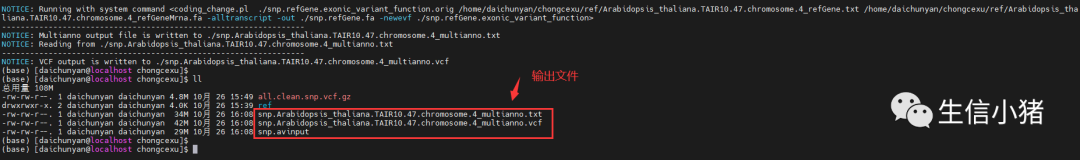
1)all.clean.snp.vcf.gz:我们要注释的vcf文件
2) /home/daichunyan/chongcexu/ref:上面构建数据库输出的两个文件Arabidopsis_thaliana.TAIR10.47.chromosome.4_refGeneMrna.fa和Arabidopsis_thaliana.TAIR10.47.chromosome.4_refGene.txt所在路径,因为 table_annovar.pl 脚本注释时就要读取这两个文件。
3)-buildver Arabidopsis_thaliana.TAIR10.47.chromosome.4:表示构建数据库输出的两个文件_refGeneMrna.fa和_refGene.txt文件的前缀。
4) --outfile ./snp : 指定输出文件前缀为snp,结果文件将以该前缀开始,输出到当前目录下。
5) -remove : 表示删除中间文件
6) -protocol : 后跟注释来源数据库名称,每个protocal名称或注释类型之间只有一个逗号,并且没有空白
7) -operation : 后跟指定的注释类型,和protocol指定的数据库顺序是一致的,g代表gene-based、r代表region-based、f代表filter-based
8) -nastring . : . 在注释结果中用于表示缺失值的字符串。在这里,. 表示缺失值。
9) -vcfinput :将处理 VCF 格式的输入数据,并生成 VCF 格式的注释结果文件
注释完这个annovar,会输出两个重要的文件。一个是 TXT(snp.Arabidopsis_thaliana.TAIR10.47.chromosome.4_multianno.txt)文件,一个就是vcf(snp.Arabidopsis_thaliana.TAIR10.47.chromosome.4_multianno.vcf)文件。snp.avinput这个文件就是一个中间文件,就是没有什么用了,大家如果不想要它就是可以直接删掉。
ANNOVAR注释完之后,就会在我们的这个结果里面添加五列信息。下面是snp.Arabidopsis_thaliana.TAIR10.47.chromosome.4_multianno.txt文件的内容
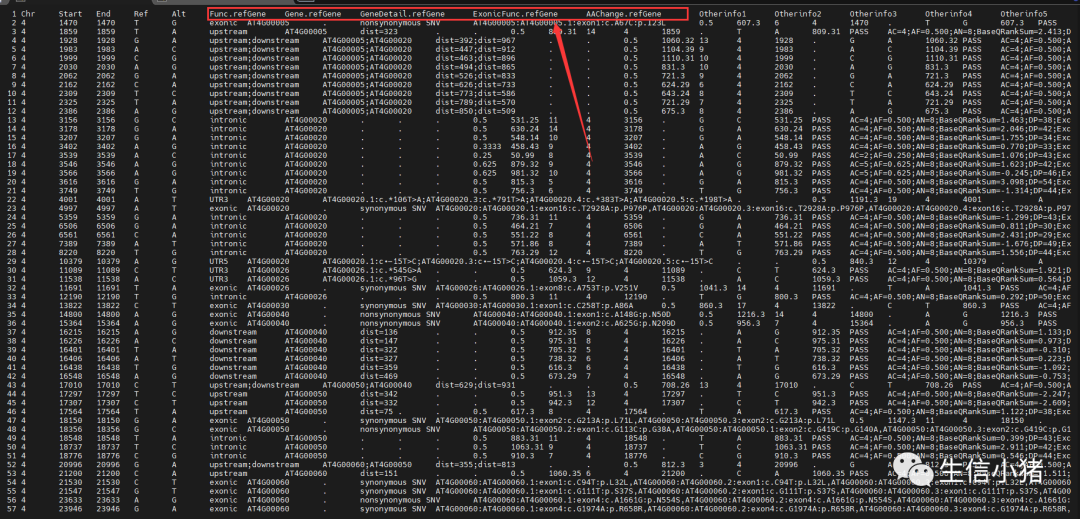
Func.refGene、Gene.refGene、GeneDetail.refGene、ExonicFunc.refGene、AAChange.refGene、Otherinfo这几列就是annovar添加的Otherinfo是样本的基因型信息,这里重点关注Func.refGene、Gene.refGene、GeneDetail.refGene、ExonicFunc.refGene、AAChange.refGene这几列。
还是把snp.Arabidopsis_thaliana.TAIR10.47.chromosome.4_multianno.txt文件复制到excel看一下吧
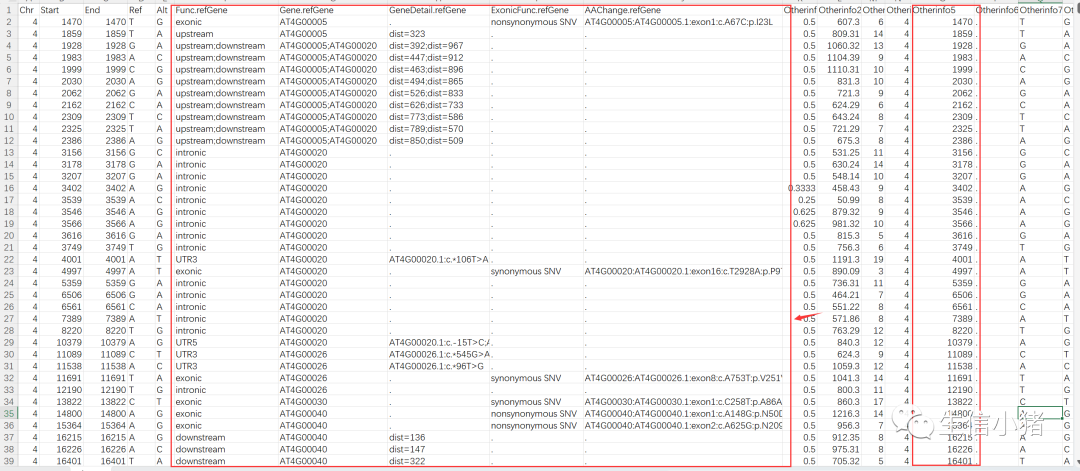
这5列的意义
1)Func.refGene:对变异位点所在的区域进行注释(exonic, splicing, UTR5, UTR3, intronic, ncRNA_exonic, ncRNA_intronic, ncRNA_UTR3, ncRNA_UTR5, ncRNA _splicing, upstream, downstream, intergenic)
2)Gene.refGene:列出该变异位点相关的转录本(只有功能符合 Func 列的转录本才列出)。如果 Func 为intergenic,此处列出两侧的基因名。
3)GeneDetail.refGene:描述 UTR、splicing、ncRNA_splicing 或 intergenic 区域的变异情况。当 Func 列的值为exonic、ncRNA_exonic、intronic、ncRNA_intronic、upstream、downstream、upstream;downstream、ncRNA_UTR3、ncRNA_UTR5 时,该列为空;当 Func 列的值为 intergenic 时,该列格式为dist=1366;dist=22344,表示该变异位点距离两侧基因的距离。
4)ExonicFunc.refGene:外显子区的 SNV or InDel 变异类型(SNV 的变异类型包括 synonymous_SNV, missense_SNV, stopgain_SNV, stopgloss_SNV 和 unknown;Indel 的变异类型包括 frameshift insertion, frameshift deletion, stopgain, stoploss, nonframeshift insertion, nonframeshift deletion 和 unknown)
5)AAChange.refGene:氨基酸改变,只有当 Func 列为 exonic 或 splicing 时,该列才有结果。按照每个转录本进行注释。
例如,NADK:NM_001198995:exon10:c.1240_1241insAGG:p.G414delinsEG,
1)NADK 表示该变异所在的基因名称,
2)NM_001198995 表示该变异所在的转录本 ID,
3)exon10 表示该变异位于转录本的第 10 个外显子上,
4)c.1240_1241insAGG表示该变异引起 cDNA 在第 1240 和 1241 位之间插入 AGG,
5)p.G414delinsEG 表示该变异引起蛋白序列在第 414 位上的氨基酸由 Gly 变为 Gly-Glu。
再如, FMN2:NM_020066:exon1:c.160_162del:p.54_54del,表示该变异引起 cDNA 的第 160 到 162 位发生删除,p.54_54del 表示该变异引起蛋白序列在第 54 位上的氨基酸删除)。
这5列的意义这部分内容来自帖子https://www.omicsclass.com/article/464,更加详细的内容可以自行查看。
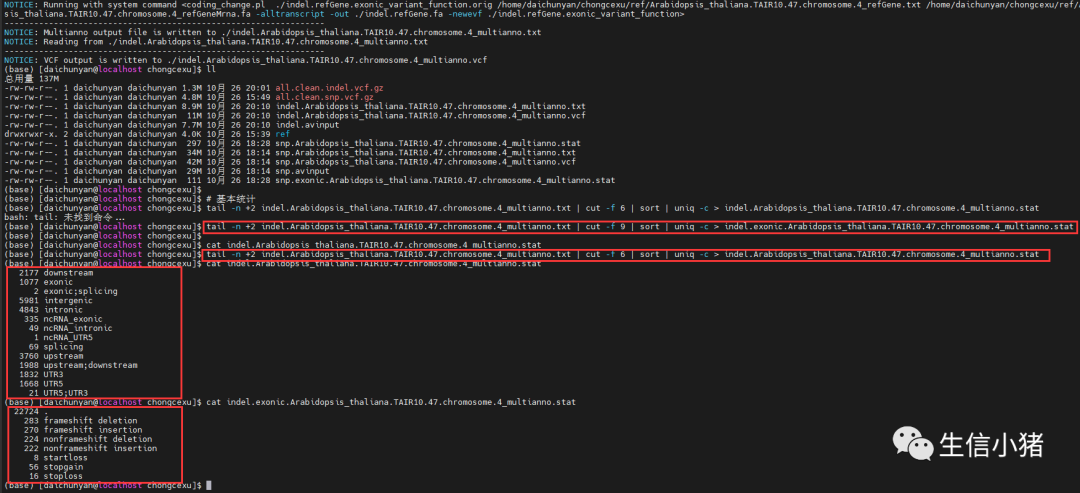
进行基本统计
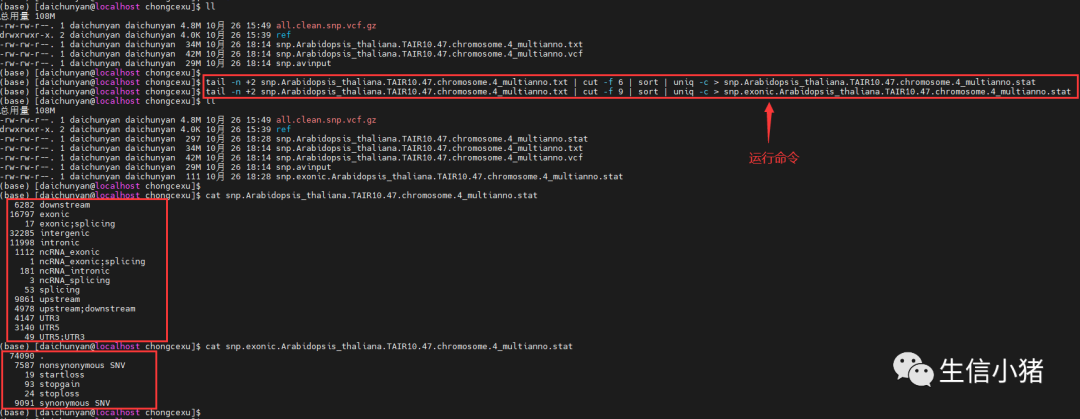
tail -n +2 snp.Arabidopsis_thaliana.TAIR10.47.chromosome.4_multianno.txt | cut -f 6 | sort | uniq -c > snp.Arabidopsis_thaliana.TAIR10.47.chromosome.4_multianno.stattail -n +2 snp.Arabidopsis_thaliana.TAIR10.47.chromosome.4_multianno.txt | cut -f 9 | sort | uniq -c > snp.exonic.Arabidopsis_thaliana.TAIR10.47.chromosome.4_multianno.stat
2.2 注释INDEL的vcf文件
indel注释和SNP注释一样,只是换了一个输入文件
table_annovar.pl all.clean.indel.vcf.gz \/home/daichunyan/chongcexu/ref \-buildver Arabidopsis_thaliana.TAIR10.47.chromosome.4 \--outfile ./indel \-remove \-protocol refGene \-operation g \-nastring . \-vcfinput# 基本统计tail -n +2 indel.Arabidopsis_thaliana.TAIR10.47.chromosome.4_multianno.txt | cut -f 6 | sort | uniq -c > indel.Arabidopsis_thaliana.TAIR10.47.chromosome.4_multianno.stattail -n +2 indel.Arabidopsis_thaliana.TAIR10.47.chromosome.4_multianno.txt | cut -f 9 | sort | uniq -c > indel.exonic.Arabidopsis_thaliana.TAIR10.47.chromosome.4_multianno.stat
三、使用annotate_variation.pl一次注释一个数据库
3.1 注释SNP的vcf文件
3.1.1 生成表格格式输入文件
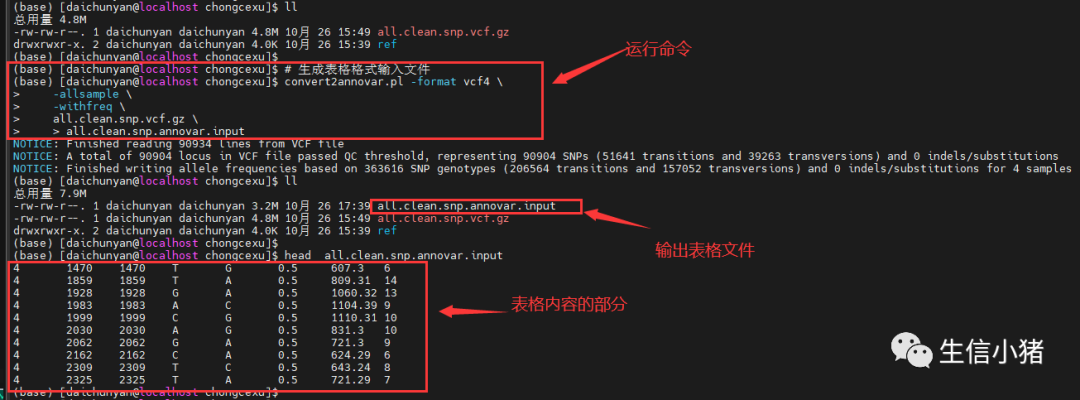
# 生成表格格式输入文件convert2annovar.pl -format vcf4 \-allsample \-withfreq \all.clean.snp.vcf.gz \> all.clean.snp.annovar.input
1)-format vcf4:输入文件格式
2)-allsample:allsample模式
3)-withfreq:输出alt频率
4)all.clean.snp.vcf.gz :输入文件
5)all.clean.snp.annovar.input:输出文件
因为输入文件并不一定需要vcf文件,软件需要的只是染色体加上坐标即可,也就是输入文件只需要是bed格式即可。其实下一步变异注释只需要5列(1)染色体位置;(2)起始位点;(3)终止位点;(4)参考基因组碱基;(5)突变碱基。即虽然将vcf转换成8列的all.clean.snp.annovar.input表格文件,但实际上下一步变异注释只用到该文件的前5列内容,这里我使用上面命令转成8列的文件,也是可以的,到时候下一步变异注释的时候也会全部输出到输出文件里。
3.1.2 变异注释
这里的注释主要有三种方式,分别是:
1)基于基因的注释, exonic,splicing,ncRNA,UTR5,UTR3,intronic,upstream,downstream,intergenic,使用 geneanno 子命令。
2)基于区域的注释, cytoBand,TFBS,SV,bed,GWAS,ENCODE,enhancers, repressors, promoters ,使用regionanno 子命令。只考虑位点坐标
3)基于数据库的过滤, dbSNP,ExAC,ESP6500,cosmic,gnomad,1000genomes,clinvar 使用 filter 子命令。考虑位点坐标同时关心突变碱基情况。
这里我们进行基于基因的注释,使用geneanno 子命令
# 进行变异注释,如果需要所有信息,添加-separateannotate_variation.pl \-buildver ./ref/Arabidopsis_thaliana.TAIR10.47.chromosome.4 \-outfile ./snp \-dbtype refGene \-geneanno \--neargene 2000 \-exonsort \all.clean.snp.annovar.input \./
1)-buildver ./ref/Arabidopsis_thaliana.TAIR10.47.chromosome.4:表示构建数据库输出的两个文件_refGeneMrna.fa和_refGene.txt文件的前缀。
2)-outfile ./snp:指定输出文件前缀为snp,结果文件将以该前缀开始,输出到当前目录下。
3)-dbtype refGene:后跟注释来源数据库名称,每个protocal名称或注释类型之间只有一个逗号,并且没有空白
4)-geneanno:基于基因进行注释
5)--neargene 2000:确定基因上下游范围
6)-exonsort:结果按照exon进行排序
7)all.clean.snp.annovar.input:输入文件
8)./:输入数据库路径
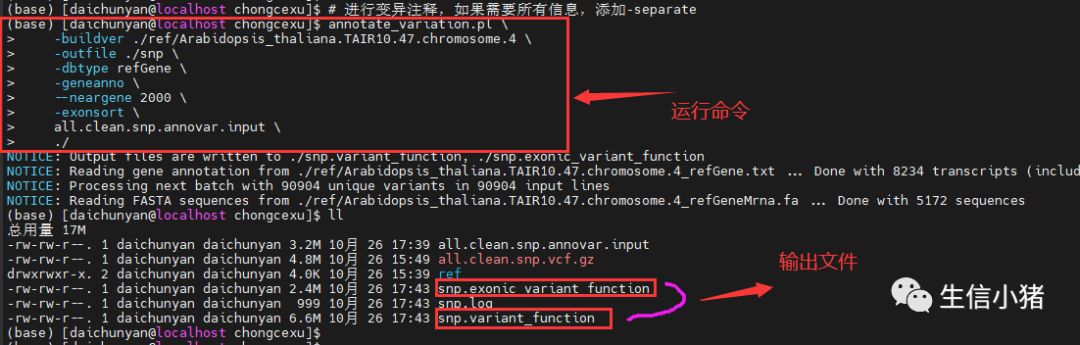
输出3个文件snp.variant_function,snp.exonic_variant_function和snp.log
查看snp.variant_function文件(把文件内容复制到excel去看)
具体可以见官网说明:http://annovar.openbioinformatics.org/en/latest/user-guide/gene/#output-file-2-refseq-gene-annotation
snp.variant_function注释所有变异所在基因及位置。所有的结果是通过在文件最前面加入2列,以tab分割,第1列为变异所在的类型,如外显子等,第2列是对应的基因名(若有多个基因名用,隔开),从第3列开始及后面的所有列其实是输入文件内容(all.clean.snp.annovar.input表格里面的内容)。
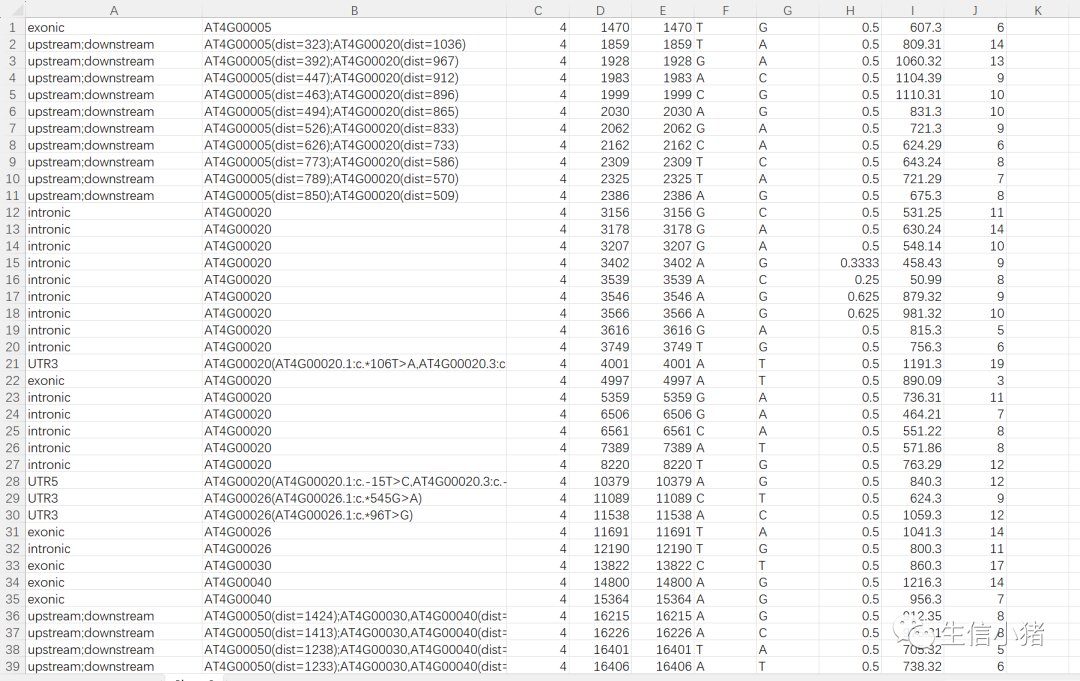
查看snp.exonic_variant_function文件(把文件内容复制到excel去看)
snp.exonic_variant_function详细注释外显子区域的变异功能、类型、氨基酸改变等。所有的结果是通过在文件最前面加入3列,以tab分割,第1列为.variant_function文件中该变异所在行号,第2列为变异功能性后果,如外显子改变导致的氨基酸变化,阅读框移码,无义突变,终止突变等,第3列包括基因名称、转录识别标志和相应的转录本的序列变化,从第4列开始及后面的所有列其实是输入文件内容(all.clean.snp.annovar.input表格里面的内容)。
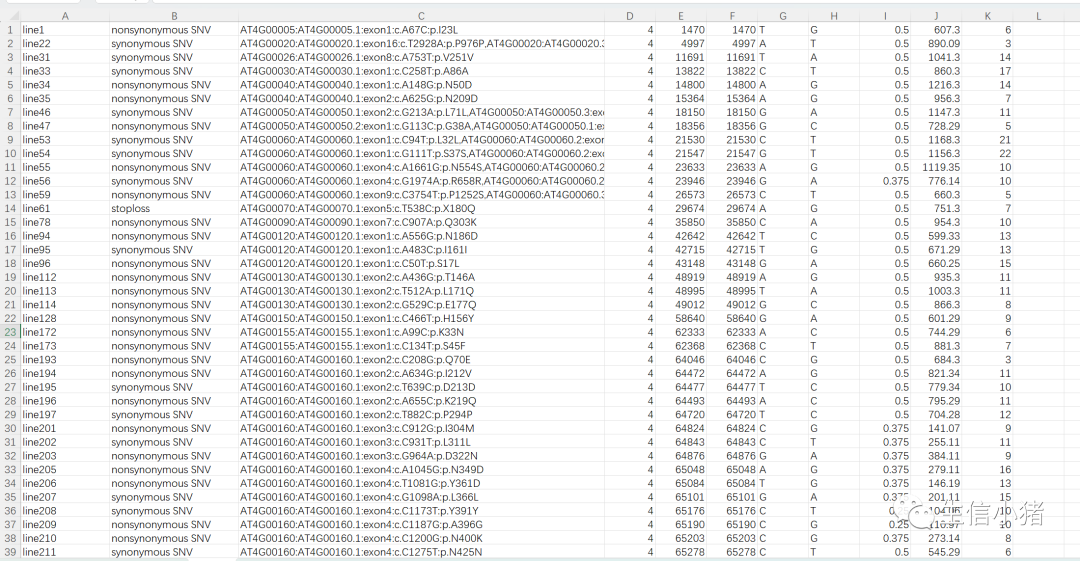
进行基本统计
cut -f 2 snp.exonic_variant_function | sort | uniq -c > snp.exonic_variant_function.statcut -f 1 snp.variant_function | sort | uniq -c > snp.variant_function.stat
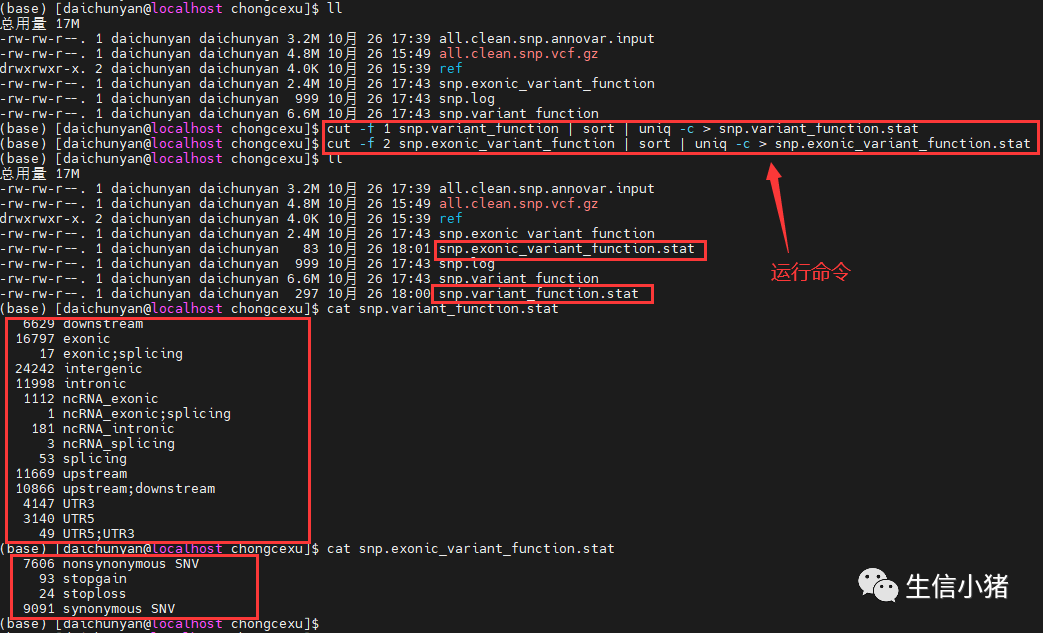
在两种方法进行基本统计时发现 snp.Arabidopsis_thaliana.TAIR10.47.chromosome.4_multianno.stat和snp.variant_function在downstream,intergenic,upstream,upstream;downstream不一样,分别把两个文件的这几个值加起来,能发现两个文件这几个值的总和是一样的。
是因为使用table_annovar.pl 一次注释多个数据库方法时,默认是确定基因上下游范围1000 bp,而使用annotate_variation.pl一次注释一个数据库时我把--neargene参数设置成2000,即确定基因上下游范围2000 bp。如果在使用annotate_variation.pl注释时把annotate_variation.pl参数设置成1000,两个就保持一致了。
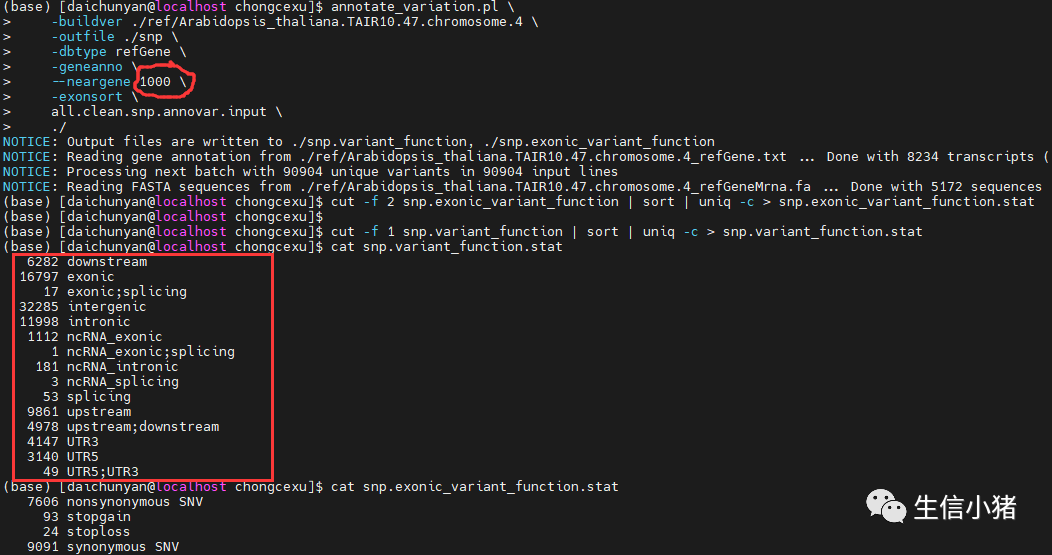
在对indel的vcf文件进行注释时,我把--neargene参数设为 1000,两个的结果也是一致的。
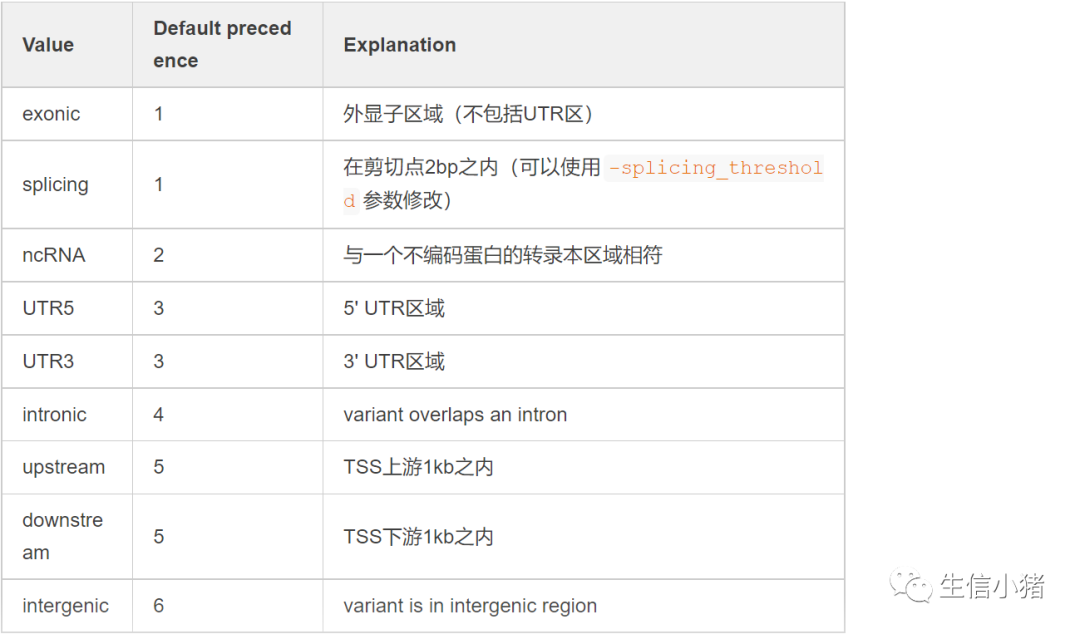
variant_function注释,优先级排序为exonic = splicing > ncRNA > UTR5/UTR3 > intron > upstream/downstream > intergenic
3.2 注释INDEL的vcf文件
# 生成表格格式输入文件convert2annovar.pl -format vcf4 \-allsample \-withfreq \all.clean.indel.vcf.gz \> all.clean.indel.annovar.input# 进行变异注释,如果需要所有信息,添加-separateannotate_variation.pl \-buildver ./ref/Arabidopsis_thaliana.TAIR10.47.chromosome.4 \-outfile ./indel \-dbtype refGene \-geneanno \--neargene 1000 \-exonsort \all.clean.indel.annovar.input \./# 基本统计cut -f 2 indel.exonic_variant_function | sort | uniq -c > indel.exonic_variant_function.statcut -f 1 indel.variant_function | sort | uniq -c > indel.variant_function.stat
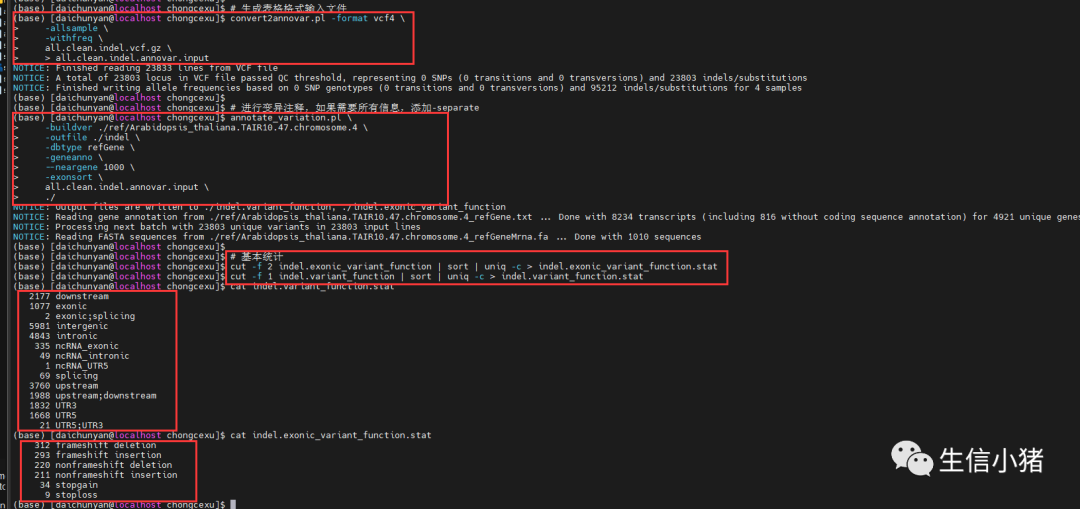
可以根据需要后续分析选择使用table_annovar.pl 一次注释多个数据库还是选择使用annotate_variation.pl一次注释一个数据库。
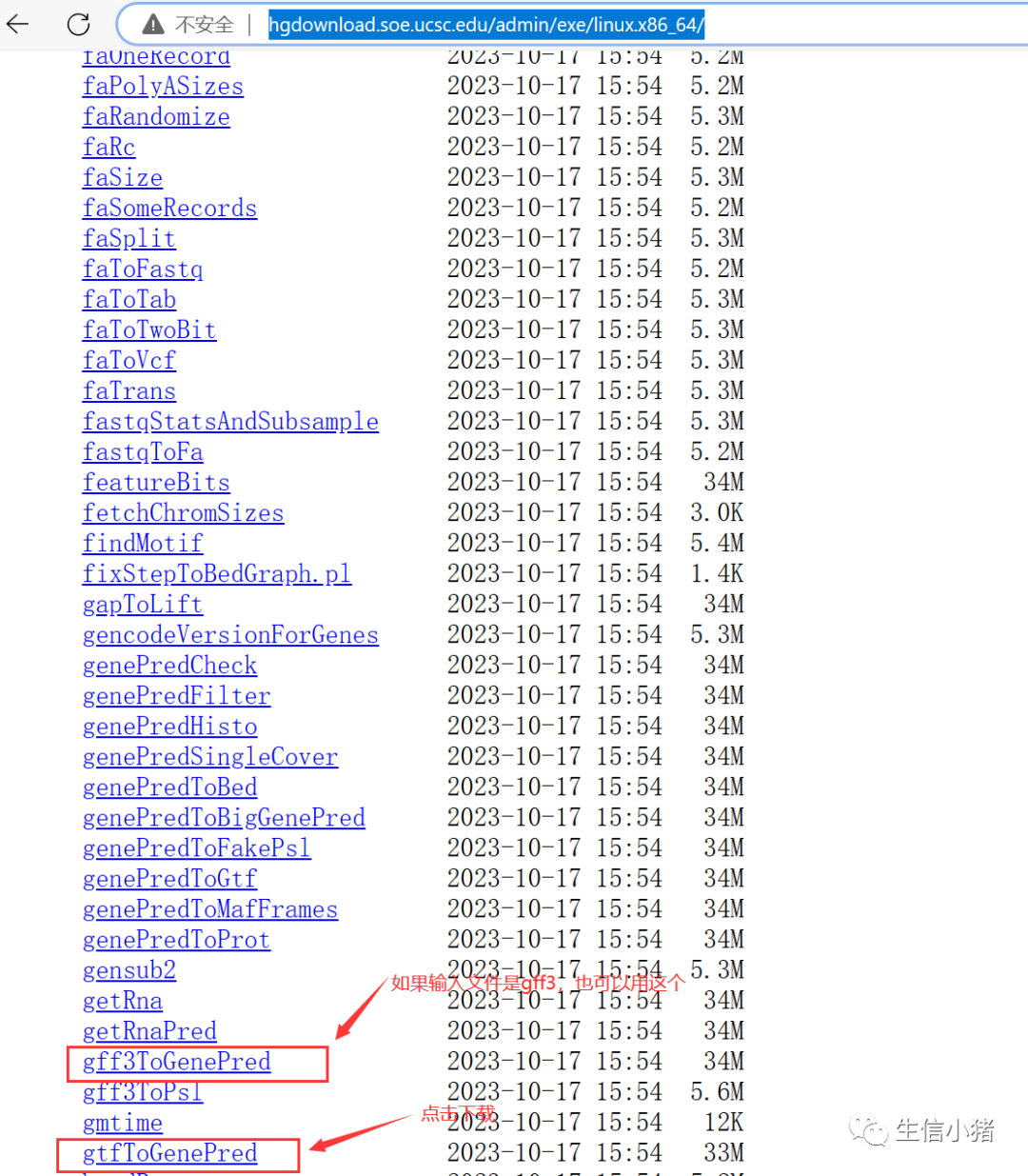
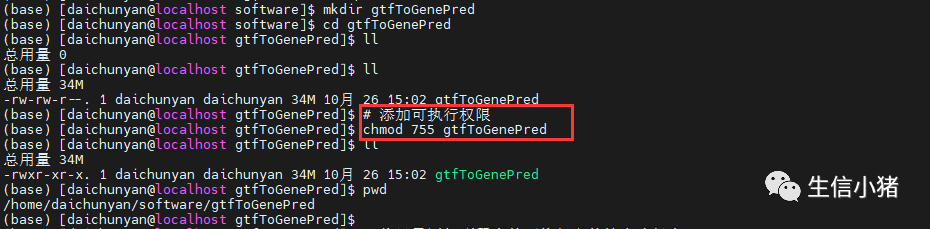
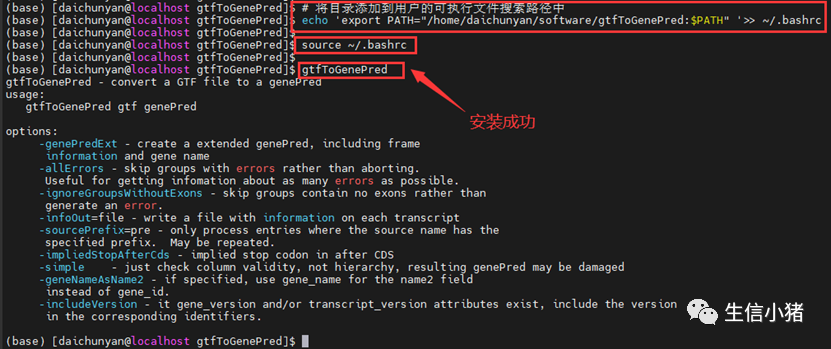
补充1、gtfToGenePred软件的安装
官网地址:http://hgdownload.soe.ucsc.edu/admin/exe/linux.x86_64/

















 457
457

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









