






原创 微信公众号生信小猪 生信小猪 2024-04-27 22:38 陕西
本次脚本的主要功能是将VCF格式文件转换为HapMap格式的文件。
VCF(Variant Call Format)是一种常用的基因组学数据格式,用于记录基因变异,包括单核苷酸多态性(SNP)和其他类型的基因突变。
HapMap是一种广泛使用的基因组数据格式,通常用于遗传学研究和关联研究。由于两者的结构和用途不同,转换VCF到HapMap对于某些遗传学和基因组学分析非常有用。
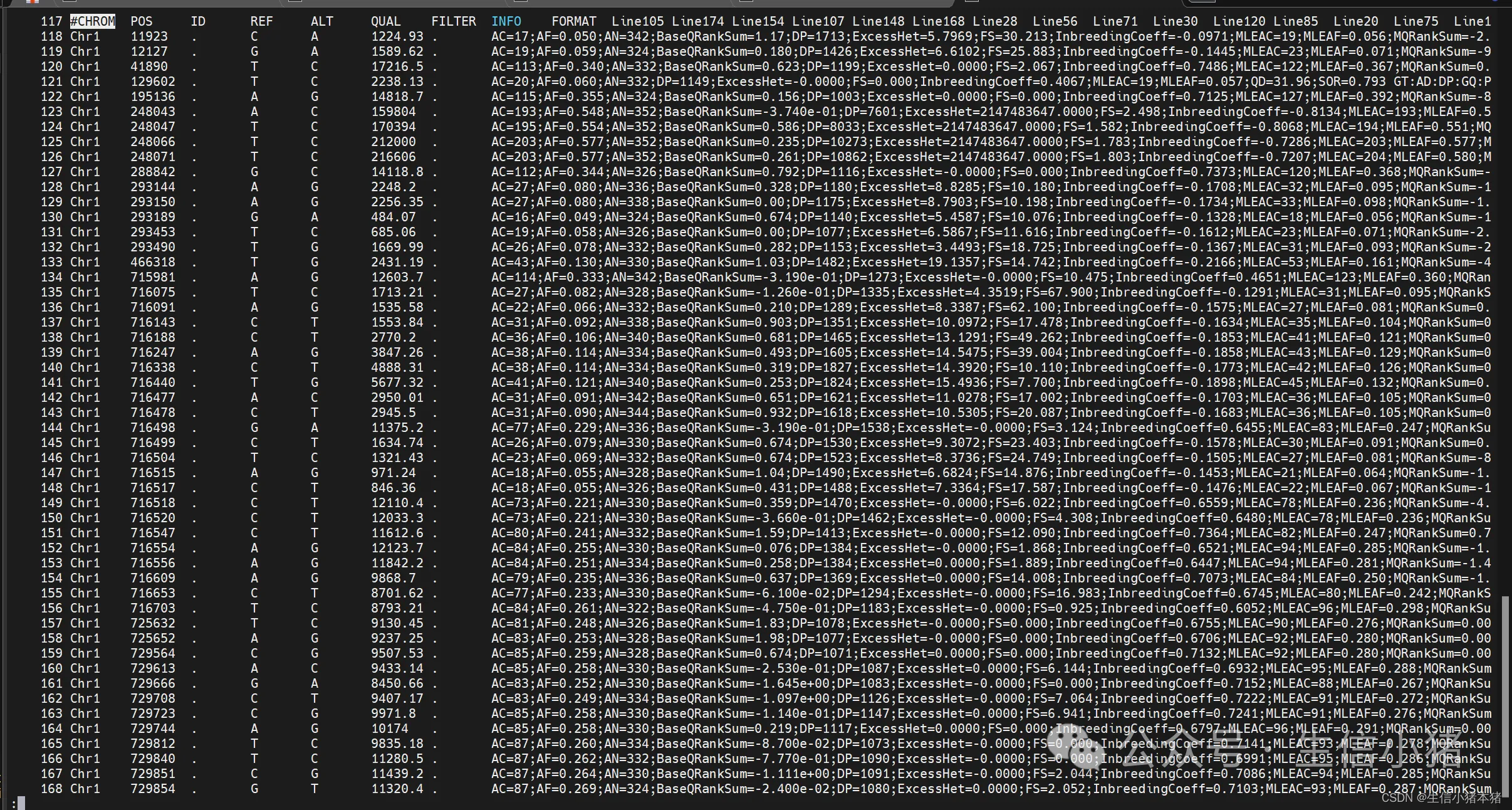
一、使用到的数据,all.clean.vcf.gz文件
下面是all.clean.vcf.gz文件的部分内容
二、在linux中运行
下面是在linux下具体运行convert_vcf_to_hapmap.py步骤
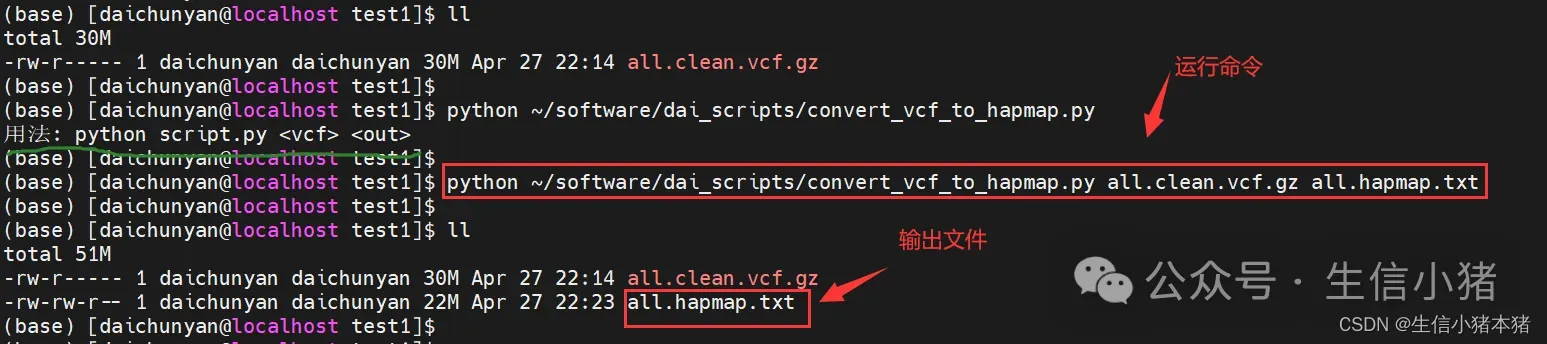
运行命令解释
1)python:这是运行Python程序的命令。
2)~/software/dai_scripts/convert_vcf_to_hapmap.py:这是脚本的路径。~ 代表用户的家目录,整个路径指向 convert_vcf_to_hapmap.py 脚本。
3)all.clean.vcf.gz:这是输入文件的路径和名称,表示一个压缩的VCF文件。
4)all.hapmap.txt:这是输出文件的路径和名称,表示转换后的HapMap格式数据将被写入这个文件。
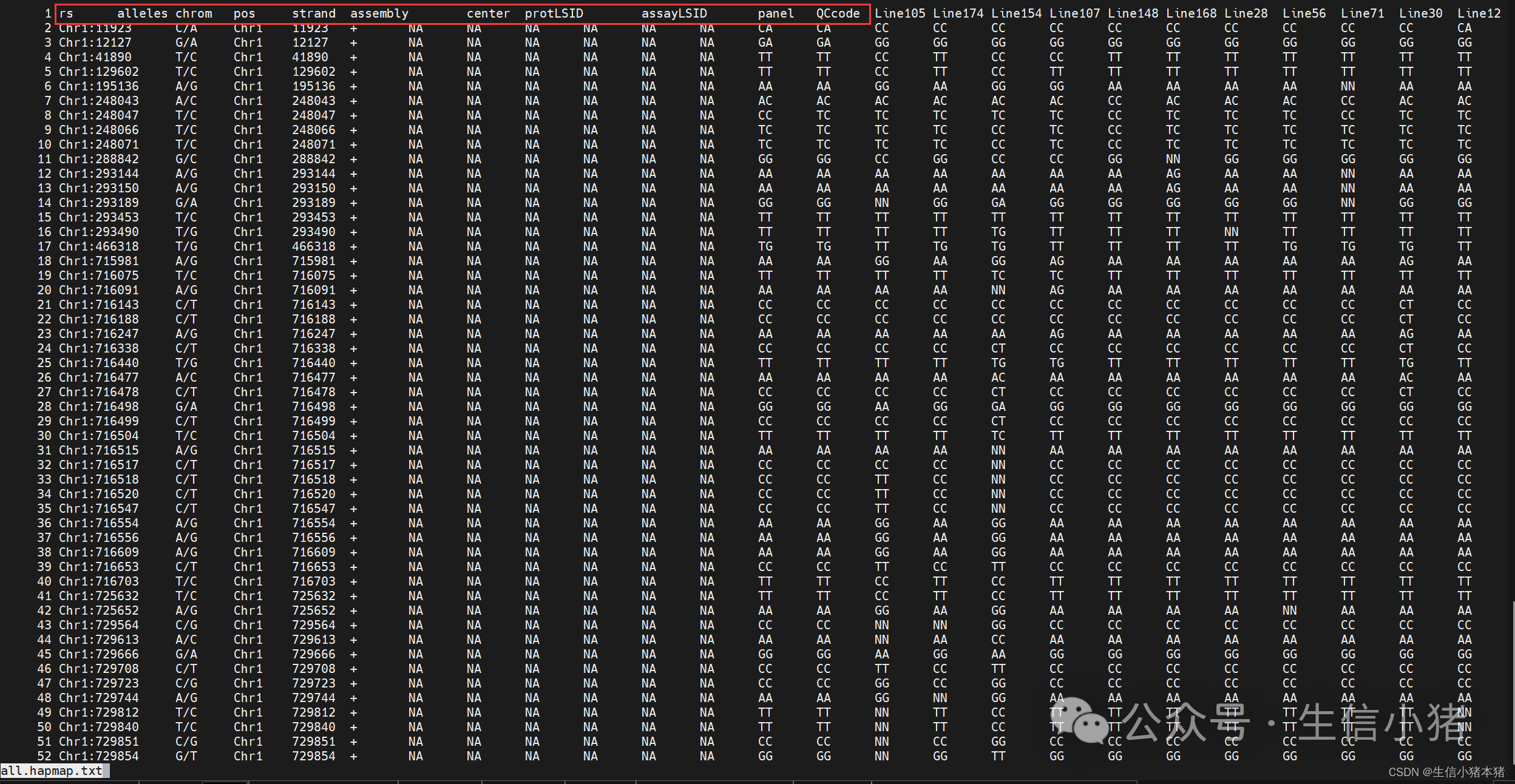
下面是输出文件all.hapmap.txt
下面是convert_vcf_to_hapmap.py脚本的全部内容
import gzipimport sys# 检查输入参数的数量,确保提供了输入和输出文件名if len(sys.argv) != 3:sys.exit("用法: python script.py <vcf> <out>")# 获取输入和输出文件名vcffile = sys.argv[1]outfile = sys.argv[2]# 根据文件扩展名,选择合适的文件打开方式if vcffile.endswith("gz"):infile = gzip.open(vcffile, "rt") # "rt"表示以文本模式读取gzip文件else:infile = open(vcffile, "r")if outfile.endswith("gz"):outfile = gzip.open(outfile, "wt") # "wt"表示以文本模式写入gzip文件else:outfile = open(outfile, "w")# 初始化变量headerline = []is_header_found = False #这行代码初始化了一个名为 is_header_found 的布尔变量,并赋予了 False 的初始值。在这里,is_header_found 变量用于标记是否已经找到了标题行。初始时,将其设置为 False 表示程序尚未找到标题行。一旦找到标题行,这个变量就会被设置为 True,以便程序知道可以开始处理数据部分了。# 循环读取输入文件for line in infile:line = line.strip() # 去掉行尾的空白字符# 检查是否是标题行if line.startswith("#CHROM"):is_header_found = Trueheaderline = line.split("\t") # 分割标题行# 写入HapMap文件的标题部分outfile.write("rs\talleles\tchrom\tpos\tstrand\tassembly\tcenter\tprotLSID\tassayLSID\tpanel\tQCcode")for i in range(9, len(headerline)): # 处理样本信息outfile.write("\t" + headerline[i])outfile.write("\n") # 新行# 如果该行不是以 "#" 开头,并且已经找到了标题行,则继续执行代码块中的操作。if not line.startswith("#") and is_header_found:splitline = line.split("\t")if len(splitline[3]) == 1 and len(splitline[4]) == 1: # 只处理单核苷酸多态性。检查了两个条件,确保该行中的REF和ALT(参考与替代碱基)长度都是1。这表明该行对应于单核苷酸多态性(SNP),而非插入、缺失等其他类型的变异。如果两者长度都为1,表示这行代表一个SNP,程序继续处理,否则忽略该行。# 写入HapMap文件中的主要数据rs_id = f"{splitline[0]}:{splitline[1]}" # rs_id:这是位点的标识符,由染色体号和位置组成。用f-string格式化为 <染色体号>:<位置>。alleles = f"{splitline[3]}/{splitline[4]}" # alleles:这表示这个位点的参考和替代碱基,用 / 分隔。chrom = splitline[0] # 染色体号pos = splitline[1] # 位点在染色体上的位置。# 写入基因位点的基本信息outfile.write(f"{rs_id}\t{alleles}\t{chrom}\t{pos}\t+\tNA\tNA\tNA\tNA\tNA\tNA")# 写入每个样本的基因型信息# 这个循环从第9列开始迭代,直到最后一列。这通常对应于VCF文件中的样本数据列。# headerline 是标题行的数据,通常包含样本名称。for i in range(9, len(headerline)):genotype = splitline[i].split(":")[0] # 这一行代码提取了每个样本的基因型信息。VCF文件中,基因型信息通常在一个字段内,其中不同部分用:分隔。基因型通常是第一个部分,所以这里使用split(":")[0]来获取基因型。if genotype in ["0/0", "0|0"]: # 如果基因型是0/0或0|0,这表示同质的参考基因型(两个相同的参考等位基因)。因此,输出文件写入参考等位基因两次。outfile.write("\t" + splitline[3] + splitline[3])elif genotype in ["0/1", "0|1"]: # 如果基因型是0/1或0|1,这表示杂合基因型(一个参考等位基因和一个替代等位基因)。因此,输出文件写入参考和替代等位基因各一次。outfile.write("\t" + splitline[3] + splitline[4])elif genotype in ["1/1", "1|1"]: # 如果基因型是1/1或1|1,这表示同质的替代基因型(两个相同的替代等位基因)。因此,输出文件写入替代等位基因两次。outfile.write("\t" + splitline[4] + splitline[4])elif genotype in ["./.", "."]: # 如果基因型是./.或.,表示未知基因型。在这种情况下,输出文件写入"NN"以表示缺失的数据。outfile.write("\tNN") # 未定基因型else:# 如果遇到未知的基因型,抛出错误sys.exit(f"错误:在 {chrom}:{pos} 位置发现无效的基因型 {splitline[i]}")outfile.write("\n") # 行结束# 关闭文件outfile.close()infile.close()
python48
python · 目录
上一篇python--将多个文件合并成一个文件,并在最后添加文件名列下一篇根据基因在染色体的位置,把某一区间的所有基因提取出来
人划线















 1715
1715

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









