










写在前面
参考官网pysam API:https://pysam.readthedocs.io/en/latest/api.html
属性常用的一些属性,读取SAM文件、FASTA文件。
读取SAM文件
读取sam文件时,熟悉一些常用的属性。
导入pysam模块
import pysam
使用pysam读取sam/bam文件,并打印一条read信息:
mysam = '/mydir/xxx.bam'
sam_rd = pysam.AlignmentFile(mysam, 'rb') # bam文件,指定使用二进制读取
for rec in sam_rd:
print(rec)
break
sam_rd.close()
# 或用with, 就不需要写close()
with pysam.AlignmentFile(mysam, 'rb') as sam_rd:
for rec in sam_rd:
print(rec)
break
使用pysam写入一条read信息到sam/bam文件:
outsam = '/mydir/out.bam'
sam_wrt = pysam.AlignmentFile(outsam, 'wb', template=sam_rd) # wb是使用二进制写入;template模板header部分使用sam_rd中的头部header
sam_wrt.write(rec) # rec是sam_rd读取的一条record
sam_wrt.close()
# 或用with, 就不需要写close()
with pysam.AlignmentFile(outsam, 'wb', template=sam_rd) as sam_wrt:
sam_wrt.write(rec)
- 获取sam各列信息:
# 示例: print(rec.query_name) # 获取第一列,readID print(rec.flag) # 获取第二列,FALG值 print(rec.reference_name) # 获取第三列,reference名称列 说明 使用pysam属性 1 readID名 rec.query_name 2 FALG值 rec.flag 3 参考序列名称 rec.reference_name 4 比对位置 rec.reference_start; rec.pos[弃用] 5 比对质量值 rec.mapping_quality 6 CIGAR值 rec.cigarstring; rec.cigar[弃用] 7 配对read的参考序列名称 rec.next_reference_name 【对PE序列有效】 8 配对read的比对位置 rec.next_reference_start 【对PE序列有效】 9 序列模板长度 rec.template_length 10 序列 rec.query_sequence 11 对应序列的碱基质量值 rec.query_qualities; rec.qual[弃用] 之后 候选字段标签tag rec.get_tags(返回tuple类型)

# 更多相似属性:
print(rec.reference_id) # 得到的是从0开始的数值(对应是按构建fasta文件的名称顺序)
print(rec.next_reference_id) # 配对read的参考序列id,类似rec.reference_id
# print(rec.positions) # 弃用
print(rec.get_reference_positions()) # 获取比对上的碱基对应的所有位置,比如: [10,11,12,13,14]
# 这些位置是参考基因组的绝对位置,不含del信息(因为已经缺失, 所以没有其位置), 当然也没有ins
print(rec.get_reference_positions(full_length=True)) # 获取的位置信息与read长度相同,没有参考序列的位置用 None代替
# 获取与read碱基对应的位置信息使用:[(相对位置, 绝对位置, read碱基), ...]
# 注意:这里的碱基都是ref的碱基型,如果有错配使用的小写也是ref型的碱基
print(rec.get_aligned_pairs(matches_only=False, with_seq=True))
print(rec.cigarstring) # 和rec.cigar同,建议用rec.cigarstring
print(rec.cigartuples) # 获取tuple类型,比如:8S10M 对应 [(4, 8), (0, 10)]
print(rec.query_alignment_start) # 比对的相对起始位置(比如从第1bp开始比对,则为0;比如前5bp是softclip,则开始位置是从第6bp碱基开始,则这里输出位置为5;
print(rec.query_alignment_end) # 比对的相对终止位置
print(rec.query_alignment_sequence) # 比对上的序列,这里不输出softclip对应的碱基
print(rec.query_alignment_qualities) # 比对上的碱基质量值数值格式(Phred33)
print(rec.query_alignment_length) # 比对上的序列长度,包括:ins和错配(当然不包括del)
print(rec.seq) # 同rec.query_sequence,建议用rec.query_sequence
print(rec.query) # 输出的同rec.seq?
print(rec.qqual) # 输出的同rec.qual?
print(rec.query_qualities) # 序列的碱基质量数值格式(Phred33)
print(rec.query_length) # 序列长度(默认是输入序列长度,但如果在sam文件中,如果是supplementary的比对结果可能seq列(第十列)是"*",获取到的值就为0。
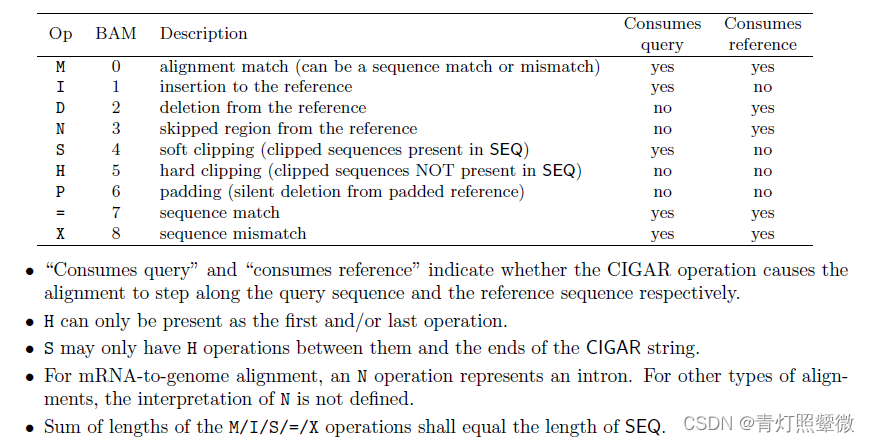
其中,rec.cigartuples中cigar值对应关系: 图来源。比如,M对应0, I对应是1,S对应4等。
-
如何将读取的信息转换成字符?
https://pysam.readthedocs.io/en/latest/api.html?highlight=tostring#pysam.AlignedSegment.to_string
print(rec) # 直接输出的bam信息,比如位置信息是0-based print(rec.tostring(sam_rd)) # 相当于直接输出为sam格式的信息(位置信息是1-based)注:
rec.tostring(sam_rd)的写法是否有问题?没问题 -
判定该记录序列信息是read1或read2,是否是反向比对?是否成对?是否合适的比对(proper paired)?
if rec.is_read1: print('rec is read1') if rec.is_read2: print('rec is read2') if rec.is_reverse: print('rec is reverse alignment') if rec.is_paired: print('rec is paired') if rec.is_proper_pair: print('rec is proper pair') -
判定该记录序列信息是否未比对上?是否是二次比对?是否是补充比对?
if rec.is_unmapped: print('rec is unmapped') if rec.is_secondary: print('rec is secondary alignment') if rec.is_supplementary: print('rec is supplementary alignment') -
配对序列是否反向比对?配对序列是否未比对上?
if rec.mate_is_reverse: print('mate read is reverse alignment') if rec.mate_is_unmapped: print('mate read is unmapped')
利用samtools view再通过FLAG值也可筛选出特定序列:flag值查询,比如用-f:samtools veiw -f256 $sam 查看二次比对的序列,使用-F是去除对应flag值的序列。

??序列unmap有两种:1)序列未map到任何位置;2)序列可map到某个位置,但由于未达到比对分值,有比对的位置,但是Cigar值为*,比如下图:一个序列,分成两段且match到了不同的方向。

读取VCF文件
读取vcf使用VariantFile,读取方式与sam类似。
import pysam
myvcf = '/mydir/xxx.vcf' # vcf.gz
vcf_rd = pysam.AlignmentFile(myvcf) # vcf文件,如果是压缩文件,增加'rb'参数,指定使用二进制读取
for rec in vcf_rd:
print(rec)
break
vcf_rd.close()
print('info', list(vcf_rd.header.info)) # 从vcf头部信息中获取,info的关键字
print('contigs', list(vcf_rd.header.contigs)) # 或者vcf文件包含的contigs,一般是染色体
print('filters', list(vcf_rd.header.filters)) # FILTER 列的可能值
print('samples', list(vcf_rd.header.samples)) # vcf文件中包含的样本名列表
vcf中,基本列信息:
| 列数 | 列名 | 解释 |
|---|---|---|
| 1 | #CHROM | 染色体 |
| 2 | POS | 位置 |
| 3 | ID | 位点ID |
| 4 | REF | ref型碱基 |
| 5 | ALT | alt型碱基 |
| 6 | QUAL | 质量值 |
| 7 | FILTER | 过滤标记 |
| 8 | INFO | 变异相关信息 |
| 9 | FORMAT | 格式名称 |
| 10 | Sample | 与格式对应值 |
每条记录record,包含的属性及对应的含义:
print(rec.chrom) # 染色体
print(rec.pos) # 位置
print(rec.ref) # ref碱基型
print(rec.alts) # alt碱基型,注意,"alts"是列表
print(rec.alts[0]) # 如果只有1个alt,使用索引0获取第一个alt型
print(rec.qual) # 变异质量值
print(list(rec.filter)) # 过滤标记
print(dict(rec.info)) # 直接转换成dict
print(rec.info.keys()) # 字典格式,获取所有keys
print(list(rec.info)) # 转成list,也可获取所有keys
print(rec.info['ABC']) # 如果有一个key是ABC,也可直接获取ABC对应的值
print(list(rec.format)) # 获取format的格式名称列表
print(rec.samples['sampleA']['DP']) # 使用sample属性,获取对应sample名的format 为DP的值
print(rec.samples[0]['DP']) # 用索引代替样本名,获取对应format为DP的值
print(rec.samples['sampleA'].values()) # 获取对应format所有信息的值,与list(rec.format)是对应的键值对
获取vcf信息还可使用bcftools工具:query命令
bcftools query -f '%CHROM\t%POS\t%REF\t%ALT\tAD:[%AD];AF:[%AF];DP:[%DP]\n' test.vcf
筛选vcf信息可以使用命令:
bcftools filter -e 'QUAL < 20 || tADA < 7 || tDPm < 100' vcf.gz
筛选INFO列,aaa或bbb对应值:
bcftools view -H -i "INFO/aaa > 0.05 && INFO/bbb <= 0.9" vcf.gz
读取FASTA文件
使用pysam处理fasta文件,示例fasta文件:
$ cat /mydir/xxx.fasta
>chr1
ATCGAG
>chr2
GCATCGAA
使用pysam读取fasta文件:
fa = '/mydir/xxx.fasta'
read_fa = pysam.FastaFile(fa)
print(read_fa.references) # fasta参考序列名称list
print(read_fa.nreferences) # fasta参考序列有几个
print(read_fa.lengths) # fasta参考序列长度
print(read_fa.fetch('chr1')) # 获取名为chr1的参考序列所有碱基
print(read_fa.fetch('chr1', 2, 5)) # 获取chr1参考序列的第3个到第5个碱基
read_fa.close()
输出内容:
['chr1', 'chr2']
2
[6, 8]
ATCGAG
CGA
注:如果没有建立索引,使用pysam会自动建立索引文件(后缀.fai) ,或使用samtools faidx /mydir/xxx.fasta 建立。
附:使用pyfaidx获取指定区域碱基序列
# -----方法1------
from pyfaidx import Fasta
hg19fa = Fasta('/your/path/Homo_sapiens_assembly19.fasta')
myseq = hg19fa['12'][25380246:25380275] # 0-based
print(myseq) # 输出该区域序列的fasta格式
# >12:25380247-25380275
# ACTGGTCCCTCATTGCACTGTACTCCTCT
print(myeq.seq) # 只输出序列
# u'ACTGGTCCCTCATTGCACTGTACTCCTCT'
print(myseq.name) # 只输出序列名(染色体号)
print(myseq.start) # 起始位置(1-based)
print(myseq.end) # 终止位置(1-based)
print(myseq.complement) # 反向互补
# -----方法2------
from pyfaidx import Faidx
hg19fa = Faidx('/your/path/Homo_sapiens_assembly19.fasta')
myseq = hg19fa.fetch('12', 25380246, 25380275) # 0-based
print(myseq) # 输出该区域序列的fasta格式
# >12:25380247-25380275
# ACTGGTCCCTCATTGCACTGTACTCCTCT
应用pysam:获取指定位置的碱基
需求: 读取sam文件时,获取每个record/每个序列中指定位置的碱基。
思路:一开始想借用record.positions和record.query_alignment_sequence获取,如下:
def get_pos_base(rec, pos):
posidx = rec.positions.index(pos) # 得到pos对应索引
return rec.query_alignment_sequence[posidx] # 得到对应位置
然而,可以这样获取的前提是:当前record中的read没有indel。确切说,只要没有ins的read便可用该方式获取。
举个例子,record信息cigar和序列是:
read1: 15M ATTCA GATGTGCGAT # 假设比对的序列起始从3,4,...,17
read2: 2S15M NNATTCA GATGTGCGAT # 假设有2bp softclip
read3: 5M1D9M ATTCA -ATGTGCGAT # 为方便看,把del处加了"-"
read4: 5M1I10M ATTCATGATGTGCGAT # 为方便看,未发生ins的序列都加了" "
使用record.positions和record.query_alignment_sequence分别得到的信息如下:
read1: [3,4,5,6,7,8,9,10,11,12,13,14,15,16,17] ATTCAGATGTGCGAT
read2: [3,4,5,6,7,8,9,10,11,12,13,14,15,16,17] ATTCAGATGTGCGAT
read3: [3,4,5,6,7, 9,10,11,12,13,14,15,16,17] ATTCA ATGTGCGAT # 注意位置列表里缺少 8(实际返回值没有空格,这里为了方便看)
read4: [3,4,5,6,7,8,9,10,11,12,13,14,15,16,17] ATTCATGATGTGCGAT # 注意:位置列表与未发生ins的read1同,但是序列是该read对应的有ins的序列
biostars 是个生信问答的好地方,大部分情况下都能找到答案: Pysam fetching reads with indels: Returns wrong positions of bases。这位提问者指出,pysam获取有indel的read的位置和碱基是错的,并给出了具体的例子。
看关键的信息:
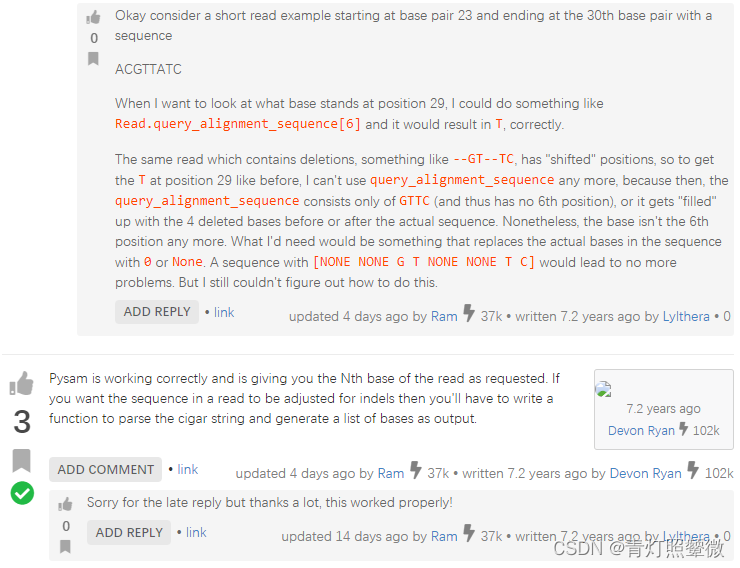
可以看出基本上,与我们的需求问题是一致的。一位回答者说是,pysam并没有提供这个需求,需要使用者结合cigar字符串写一个函数来获取。
从逻辑上看,获取碱基好像并不难。根据对应的位置,按cigar的字符和序列顺着获取碱基即可。但是在代码实现时,貌似并不是特别easy:(仍需要审核是否有遗漏没有考虑的到细节)
def query_base(seq, astr, atp, start, qpos):
"""
--------------------------------
seq: input sequence (NOTE: raw seq which match cigar, use `record.query_sequence` to get)
astr: cigar string
atp: cigar tuple
start: seq match start position
qpos: query base position
--------------------------------
return: query position base
# eg:
# seq = 'NNNNNNNNATATCCCGTGCCATATCCCGTGATATCCCGTG'
# astr = '8S10M2I20M'
# atp = [(4,8), (0, 10), (1, 2), (0, 20)]
seq = 'NNNNNNNNATATCCCGTGATATCCCGTGATATCCCGTG'
## 'NNNNNNNNATATCCCGTG ATATCCCGTGATATCCCGTG'
astr = '8S10M2D20M'
atp = [(4, 8), (0, 10), (2, 2), (0, 20)]
"""
cnt0, cnt1, cnt2 = 0, 0, 0
found_base = False
is_delbase = False
for i, j in atp:
if i == 0: # match
cnt0 += j
if i in [1, 4]: # ins, softclip
cnt1 += j
if i == 2: # del
cnt2 += j
if cnt0+cnt2 >= qpos-start+1: # ref len >= query pos len
qidx = qpos+cnt1-1-cnt2
found_base = True
if qpos-start >= cnt0: # query pos is del base
# qidx = qidx - min(qpos-start-cnt0+1, cnt2) + cnt2
# print("del pos:", qpos, qidx, qpos-start-cnt0)
is_delbase = True
break
# print(start, qpos, cnt0, cnt1, cnt2, qidx)
# print(seq[:qidx])
# print(seq)
# print('=====================')
if not found_base:
return '-1' # 位置超过查询,返回"-1"
if is_delbase:
qbase = '-' # 如果是del,返回"-"
else:
qbase = seq[qidx-1]
print(qbase)
return qbase
# eg:
# seq = 'NNNNNNNNATATCCCGTGCCATATCCCGTGATATCCCGTG'
# astr = '8S10M2I20M'
# atp = [(4,8), (0, 10), (1, 2), (0, 20)]
seq = 'NNNNNNNNATATCCCGTGATATCCCGTGATATCCCGTG'
## 'NNNNNNNNATATCCCGTG ATATCCCGTGATATCCCGTG'
astr = '8S10M2D20M'
atp = [(4, 8), (0, 10), (2, 2), (0, 20)]
query_base(seq, astr, atp, 2, 9)
query_base(seq, astr, atp, 2, 10)
query_base(seq, astr, atp, 2, 11)
query_base(seq, astr, atp, 2, 12)
query_base(seq, astr, atp, 2, 13)
query_base(seq, astr, atp, 2, 14)
query_base(seq, astr, atp, 2, 18)
query_base(seq, astr, atp, 2, 22)
附:
看到知乎上,介绍biostar:生物信息神奇网站系列(四):Biostars。之前只知道有biostarhandbook这本书:


竟然还发表了文章:BioStar: An Online Question & Answer Resource for the Bioinformatics Community

















 1190
1190

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









