






论文阅读笔记(二)——The Nucleotide Transformer: Building and Evaluating Robust Foundation Models for Human Genomics
目录
摘要
缩小可测量遗传信息和可观察性状之间的差距是基因组学长期面临的挑战。然而,仅从DNA序列预测分子表型仍然是有限和不准确的,通常是由于缺乏注释数据和无法在预测任务之间转移学习。在此,我们对DNA序列预先训练的基础模型进行了广泛的研究,命名为Nucleotide Transformer,其参数范围从50M到2.5B不等,并整合了来自3,202种不同人类基因组的信息,以及来自不同门(包括模式生物和非模式生物)的850个基因组。这些 transformer models产生可转移的,特定于上下文的核苷酸序列表示,即使在低数据设置中也可以进行准确的分子表型预测。我们表明,开发的模型可以在低成本和低可用数据制度下进行微调,以解决各种基因组学应用。尽管没有监督,变形模型学会了将注意力集中在关键的基因组元素上,包括那些调节基因表达的元素,比如增强子。最后,我们证明了利用模型表示可以提高功能遗传变异的优先级。本研究探索的基因组学基础模型的训练和应用,为从DNA序列中准确预测分子表型提供了广泛适用的垫脚石。
简介
因为太吃卡,不会复现,所以没细看
The Human reference genome dataset
人类参考数据集考虑了参考装配体GRCh38/ hg383的所有常染色体和性染色体序列,共达到32亿个核苷酸。
为了让模型了解人类自然发生的遗传多样性,我们构建了一个训练数据集,其中包括来自不同人群的遗传变异。具体来说,我们从1000个基因组计划[55]中下载了变异调用格式(VCF)文件4,该文件旨在记录人类中发生频率至少为1%的遗传变异。该数据集包含3202个高覆盖率的人类基因组,来自非洲、美洲、东亚和欧洲血统的27个地理结构人群(详见表2),总计20.5万亿核苷酸。这种多样性使得数据集能够更好地编码人类遗传变异。为了从VCF文件中进行FASTA格式的单倍型重建,我们考虑了数据的阶段性版本,共对应125M个突变,其中111M为单核苷酸多态性(snp), 14M为单核苷酸多态性(indel)。
The Multispecies dataset
为了建立一个包含大量不同基因组的数据集,我们首先分析了NCBI5上可用的基因组,然后从每个属中任意选择一个物种。植物和病毒基因组没有被考虑在内,因为它们的调控元件不同于本工作中感兴趣的那些。由此产生的基因组收集被减少到总共850个物种,其基因组加起来有1740亿个核苷酸。表3显示了每一类在数据集中核苷酸总数中所占的最终贡献(按核苷酸数量计算),与从NCBI解析的原始集合中的贡献相同。最后,我们通过选择几个在文献中被大量研究的基因组来丰富这个数据集(表4)。
DNA序列编码的依赖模式在理解基因组过程中起着重要作用,从表征调控区域到评估单个变异在其单倍型背景下的影响。最近的进展结合了卷积神经网络(CNN)和transformer架构,能够编码位于上游 100千碱基(kb) 的调控元件。
现代基因组学研究产生的大量数据既是机遇也是挑战。一方面,物种和种群间复杂的自然变异模式是现成的;另一方面,能够处理大规模数据的强大深度学习方法对于从未标记的数据集中准确提取信号是必要的。基于核苷酸序列训练的大型基础模型似乎是解决这一挑战的自然选择。
利用Nucleotide Transformer(NT Transformer),我们对如何构建和评估稳健的基础模型来编码基因组序列进行了系统的研究和基准。我们通过建立四个不同大小的不同语言模型开始了我们的研究,从500M到2.5B参数 不等。
这些模型在三个不同的数据集上进行了预训练。训练后,我们利用这些模型中的每个模型的表示(即嵌入)来同时训练它们完成18个不同的基因组策划预测任务。
为了破译在预训练中学习到的序列特征,我们探索了模型的注意图、困惑,并对它们的嵌入进行了数据降维。此外,我们通过zero-shot-based scores 评估了嵌入对人类功能重要遗传变异影响的建模能力。
在最初一组实验的基础上,我们开发了第二组四种LMs,将参数的大小从500M减少到50M,以研究这些模型的缩放规律。我们成功地构建了一个模型,该模型的性能与之前的最佳模型相匹配,同时仅使用十分之一的参数数量和两倍的感知场大小就达到了这种性能水平。我们利用引入的基准和标准化稳健评估方法,系统地将我们的8个模型与其他基础模型进行比较,一致地证明我们的最佳模型优于它们(图1a)。
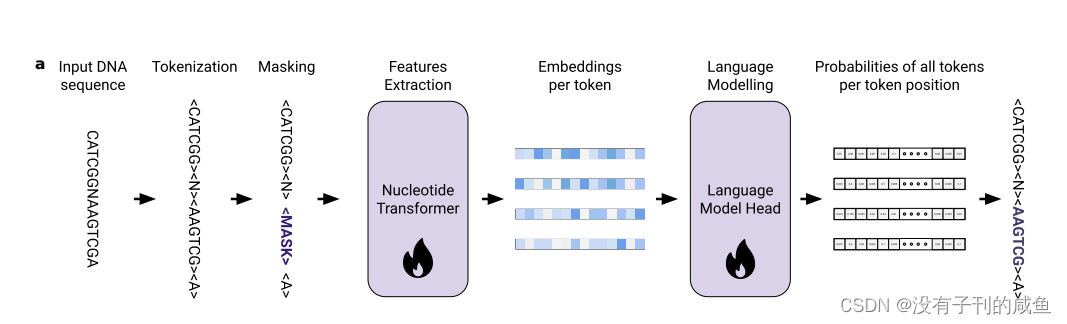
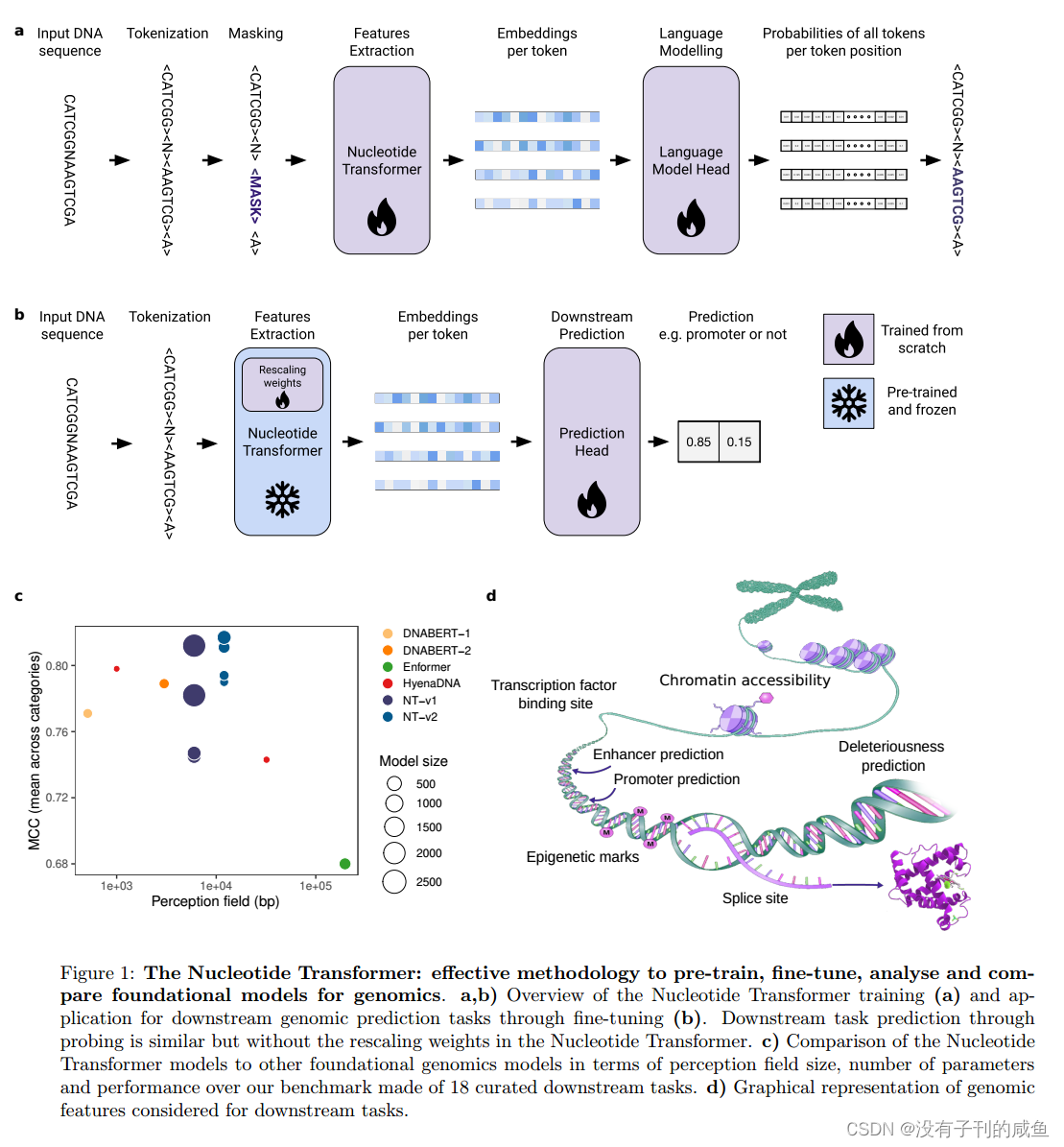
模型结构
训练LMs任务模型来预测序列中掩码位置上最可能的标记的一种技术,通常称为掩码语言建模(masked language modelling,MLM)。受MLM在蛋白质研究领域获得的结果的启发,其中蛋白质被视为句子,氨基酸被视为单词,我们将MLM应用于基因组学中训练语言模型转换器,将核苷酸序列视为句子,将k-mers (k=6)视为单词。它们由一个初始嵌入层组成,该层将输入序列中的位置转换为嵌入向量,然后是一堆自关注层,这些自关注层依次对这些嵌入进行细化。
用MLM训练语言模型转换器的主要技术被称为transformer的双向编码器表示(BERT)。在BERT中,序列中的所有位置都可以相互关注,从而允许信息在两个方向上流动,这在DNA序列的背景下是必不可少的。在训练期间,网络的最终嵌入被馈送到语言模型头部,该头部将其转换为输入序列上的概率分布。
嵌入层将标记序列转换为嵌入序列。然后将位置编码添加到序列中的每个嵌入中,以向模型提供位置信息。我们使用一个可学习的位置编码层,它最多接受1000个标记。我们使用6-mer令牌作为序列长度(最多6kb)和嵌入大小之间的权衡,因为与其他令牌长度相比,它实现了最高的性能。然后由transformer层堆栈处理令牌嵌入。每个transformer层通过层规范化层转换其输入,然后是多头自关注层。自注意层的输出与transformer的输入通过跳接求和。然后将该操作的结果传递给一个新的层归一化层和一个具有GELU激活的两层感知器。每个模型的正面数、嵌入维数、感知器隐藏层内的神经元数和总层数见表1。在自监督训练期间,堆栈的最后一层返回的嵌入由语言模型头转换为序列中每个位置上现有标记的概率分布。
我们的第二版Nucleotide Transformer v2模型包括一系列被证明更有效的架构变化:我们使用每个注意层使用的旋转嵌入,而不是使用学习的位置嵌入;我们使用门控线性单元,具有无偏置的闪动激活,使nlp更有效。这些改进的模型还可以接受多达2,048个标记的序列,从而产生12kbp的更长的上下文窗口。
关于训练设备
nucleotide-transformer-2.5b-multi-species model
2.5B使用 128 个 A100 80GB GPU 进行训练,有效批处理大小为 1M 令牌。
Nucleotide-transformer-500M-Human-Ref
使用 8 个 A100 80GB 进行训练,有效批处理大小为 1M 令牌。
Nucleotide-transformer-50M-Human-Ref
使用 8 个 A100 80GB 进行训练,有效批处理大小为 1M 令牌。
结果
核苷酸转换器模型在微调后准确地预测了不同的基因组任务。a)微调NT模型以及HyenaDNA、DNABERT和Enformer预训练模型跨下游任务的性能结果(MCC:马修相关系数)。DNABERT-1没有剪接受体和供体任务的值,因为它们的序列比模型可以处理的要长。误差条表示10倍交叉验证得出的2个SDs。b)微调后所有语言模型跨下游任务的MCC性能的归一化平均值(按类别划分)。c)与基线DeepSEA模型相比,Multispecies 2.5B模型在不同人类细胞和组织的DNase I超敏位点(DHS)、组蛋白标记(HMs)和转录因子位点预测上的表现。每个点代表不同基因组剖面的ROC曲线下面积(AUC)。标记了每个型号的平均AUC。d)与SpliceAI和其他剪接模型相比,Multispecies 2.5B模型在预测人类基因组剪接位点方面的表现。
e)与基线DeepSTARR模型相比,Multispecies 2.5B模型对果蝇S2细胞发育和内内事增强子活性的预测性能。

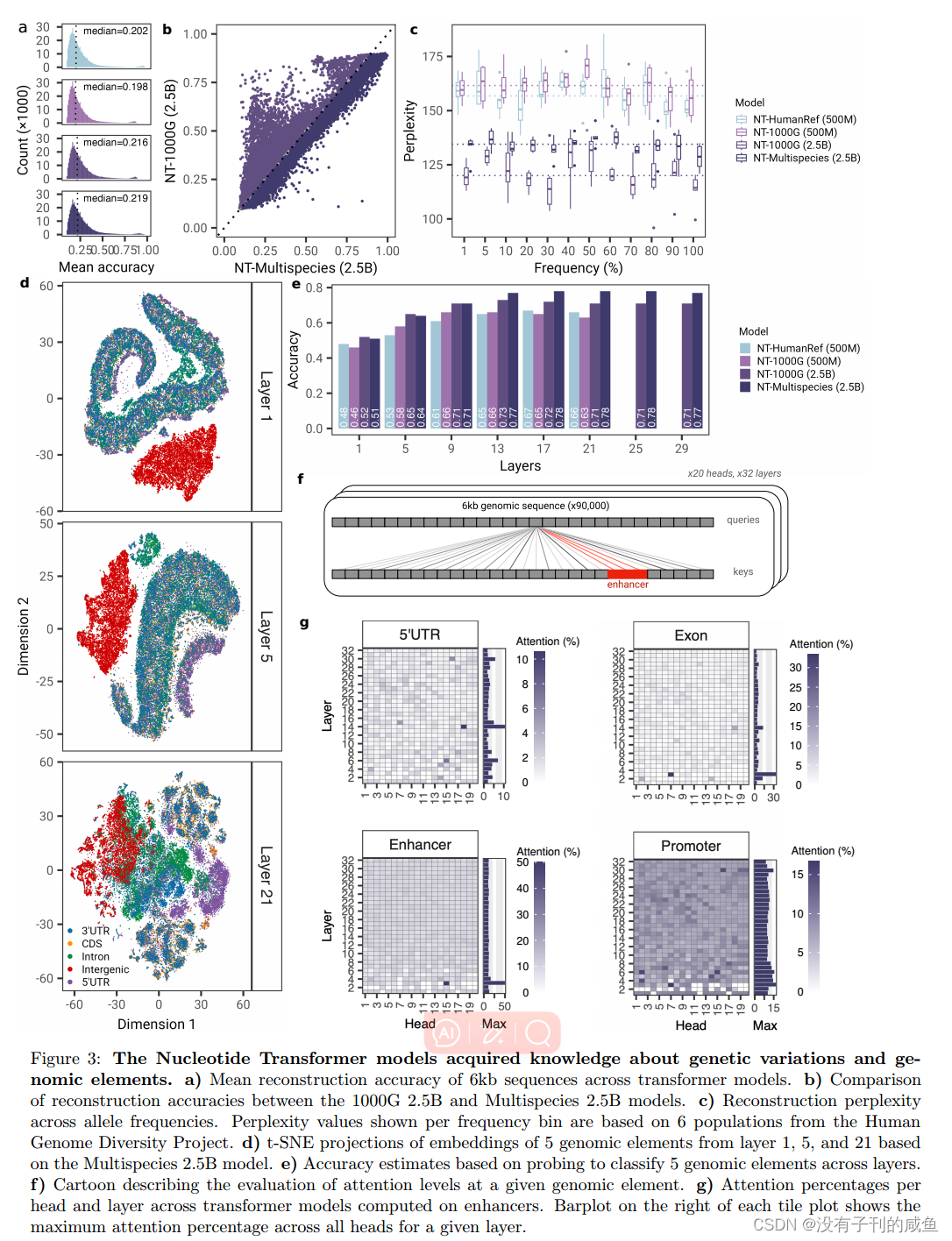
核苷酸转换器模型获得了有关遗传变异和基因组要素的知识。a)各变压器模型6kb序列的平均重建精度。b) 1000G 2.5B与Multispecies 2.5B模式重建精度比较。c)跨等位基因频率的重建困惑。每个频率箱显示的困惑值基于人类基因组多样性计划中的6个种群。d)基于Multispecies 2.5B模型的第1层、第5层和第21层5个基因组元件嵌入的t-SNE预测。e)基于探测跨层分类5个基因组元件的精度估计。
f)描述对给定基因组元素的注意水平评估的漫画。g)在增强器上计算的变压器模型的每个头和层的注意力百分比。每个平铺图右侧的条形图显示了给定图层中所有头部的最大注意力百分比。
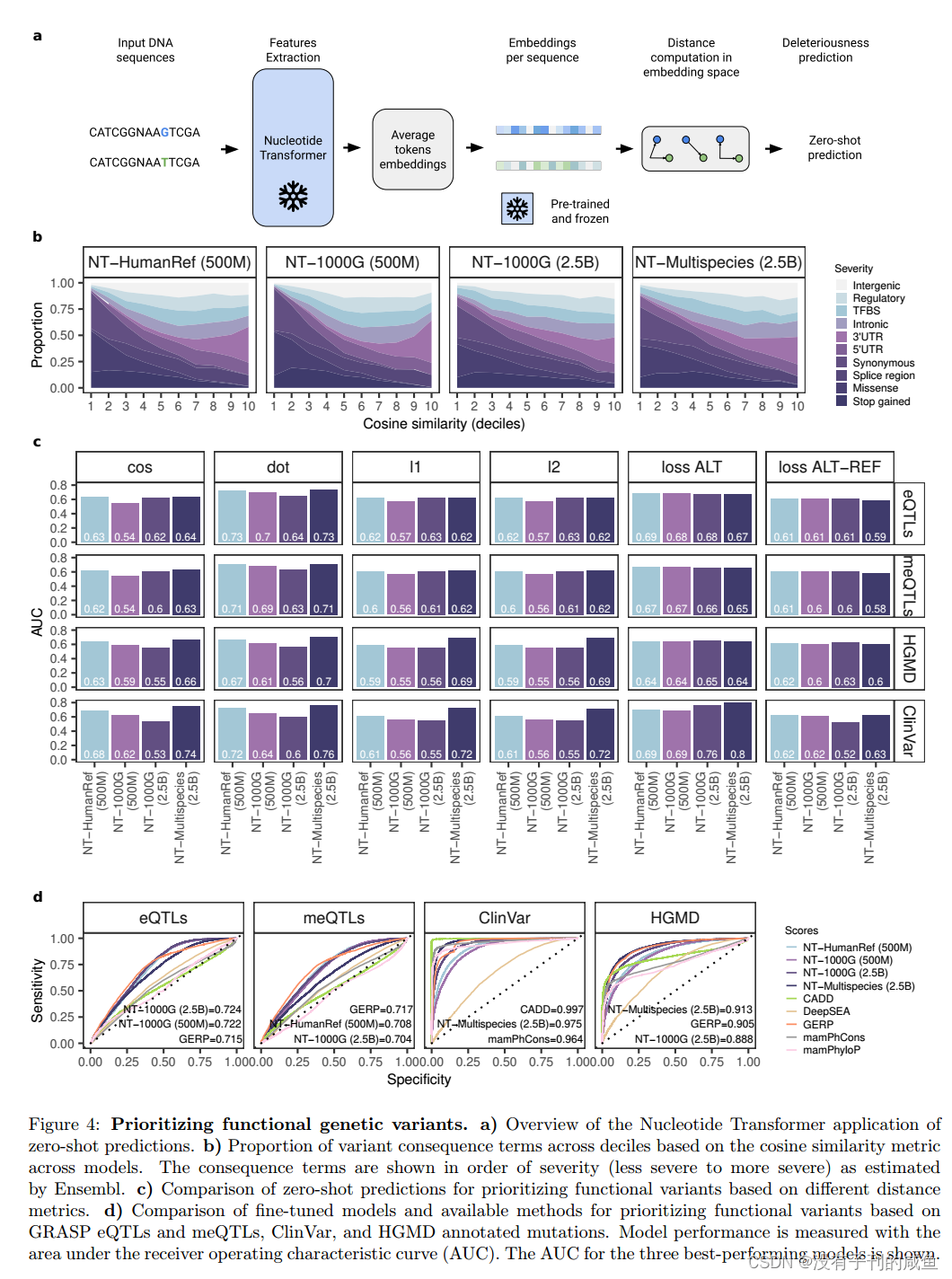
优先考虑功能性基因变异。a) Nucleotide Transformer在零概率预测中的应用概述。b)基于模型间余弦相似度度量的不同结果项在十分位数上的比例。结果项按照严重程度(从较轻到较严重)的顺序显示,正如Ensembl所估计的那样。c)基于不同距离度量的功能变量优先级的零射击预测比较。d)基于GRASP eqtl和meqtl、ClinVar和HGMD注释突变的功能变异优先排序的微调模型和可用方法的比较。模型性能用接收机工作特性曲线下面积(AUC)来衡量。给出了三种最佳表现模型的AUC。
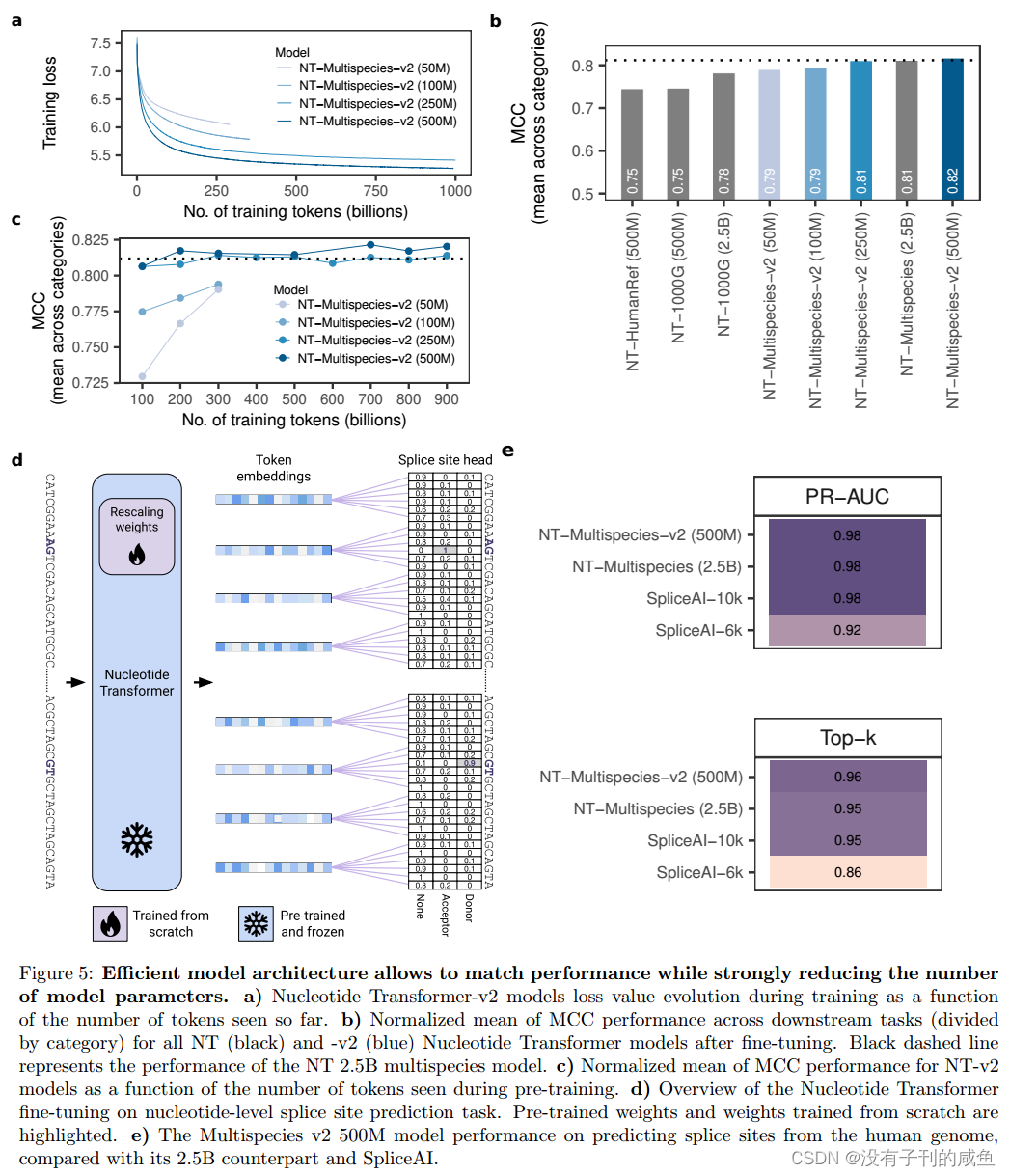
有效的模型体系结构允许匹配性能,同时大大减少模型参数的数量。a)核苷酸转换器-v2模型在训练过程中损失价值进化,作为迄今为止看到的令牌数量的函数。b)微调后,所有NT(黑色)和-v2(蓝色)Nucleotide Transformer模型在下游任务中的MCC性能的归一化平均值(按类别划分)。黑色虚线表示NT 2.5B多物种模型的性能。c) NT-v2模型的MCC性能的归一化均值作为预训练期间看到的令牌数量的函数。d)核苷酸水平剪接位点预测任务的Nucleotide Transformer微调概述。突出显示了预训练的权重和从头训练的权重。e)与25 b模型和SpliceAI相比,Multispecies v2 500M模型在预测人类基因组剪接位点方面的表现。















 1180
1180











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









