






1.fasta文件处理
1.1 seqtk
1.1.1 seq:
seqtk seq -l n(数字) file1 :每行显示n个字母
seqtk seq -L n(数字) file1 :file1 中短于n的序列删除
seqtk seq -u file1 : 小写转化为大写
1.1.2 sample
seqtk sample -s n(seed) file1 x/百分比(抽x条数据):随机抽取x条数据或百分之多少的数据
1.1.3 subseq
seqtk subseq file1 file2 :在文件1中寻找文件2的内容
1.2 seqtik
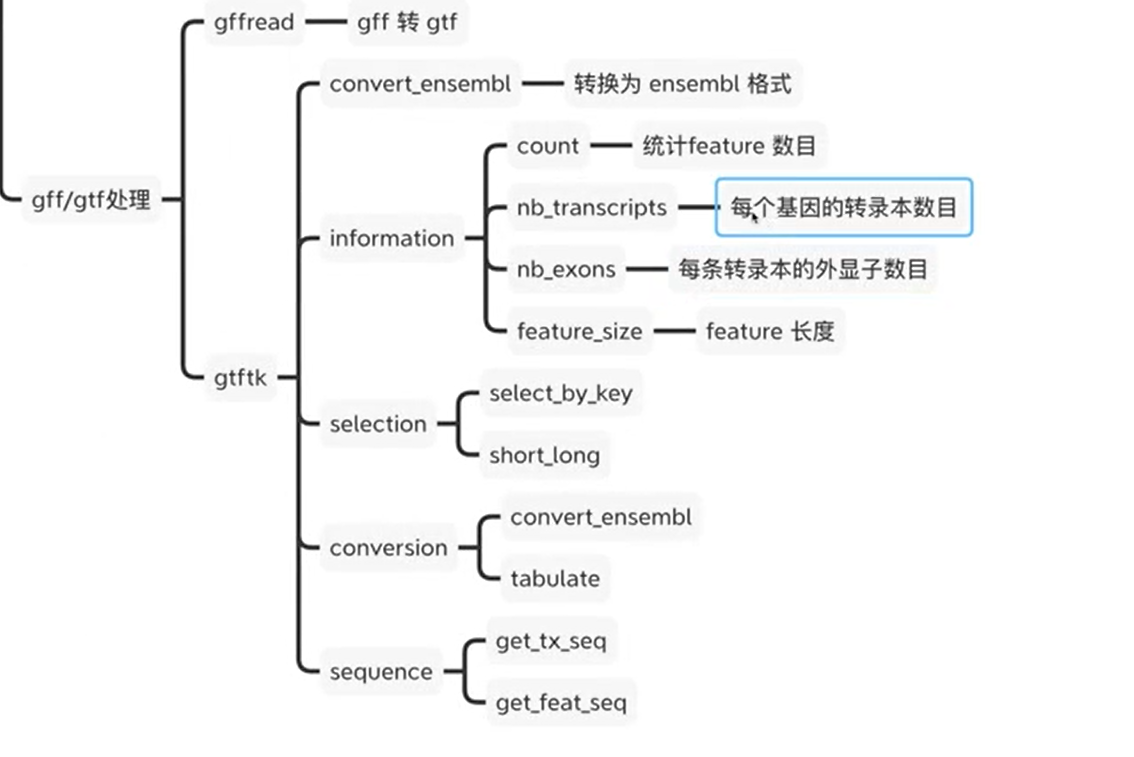
2.gtf文件处理
2.1gffread
gff转换为gtf文件
gffread -T -o file1 file2(原文件) #-o后跟输出文件名,-T表示输出必须为gtf文件
gffread -T -o genes.gtf SWO.v3.0.gene.model.gff3 #-o后跟输出文件名,-T表示输出必须为gtf文件
2.2gtftk














 90
90











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









