






############################## Star-fusion结果绘制circos图
file_fusion <- "data/cnv/result/sample1_RNA_star-fusion_res.Fusion.tsv"
file_output <- "results/01_3个病例融合图/fusion_sample1.pdf"
library(data.table)
library(stringr)
data_fusion <- fread(file_fusion)
#整理数据格式,需要的数据为:染色体号,断点位置,基因名
data_fusion$Site1_Chromosome <- data_fusion$chromosome1
data_fusion$Site1_Position <- data_fusion$softclip1
data_fusion$Site2_Chromosome <- data_fusion$chromosome2
data_fusion$Site2_Position <- data_fusion$softclip2
data_fusion$Site1_Hugo_Symbol <- str_extract(data_fusion$sclip1_info, ":(.*?):", group=1)
data_fusion$Site2_Hugo_Symbol <- str_extract(data_fusion$sclip2_info, ":(.*?):", group=1)
# 去除重复基因融合
data_fusion <- data_fusion[!duplicated(data_fusion[,c("Site1_Hugo_Symbol",
"Site2_Hugo_Symbol")]),]
# 导入绘图函数
source("R/my_functions/common_functions.R")
pdf(file=file_output,width=5,height=5)
circos_plot_fusion(data_sv_plot=data_fusion,
Site1_Chromosome="Site1_Chromosome",
Site2_Chromosome="Site2_Chromosome",
Site1_Position="Site1_Position",
Site2_Position="Site2_Position",
Site1_Hugo_Symbol="Site1_Hugo_Symbol",
Site2_Hugo_Symbol="Site2_Hugo_Symbol",
genome="hg19",
label=TRUE,
color_by_chr=FALSE,
title=paste0("Sample ID: ", file_fusion))
dev.off()
circos_plot_fusion的自定义函数代码为"R/my_functions/common_functions.R"文件中定义:
circos_plot_fusion <- function(data_sv_plot,
Site1_Chromosome="Site1_Chromosome",
Site2_Chromosome="Site2_Chromosome",
Site1_Position="Site1_Position",
Site2_Position="Site2_Position",
Site1_Hugo_Symbol="Site1_Hugo_Symbol",
Site2_Hugo_Symbol="Site2_Hugo_Symbol",
genome="hg19",
label=TRUE,
color_by_chr=FALSE,
col_red="#FF000040",
col_blue="#0000FF40",
col_grey="grey90",
color_col=NULL,
title="Circos plot"){
library(dplyr)
RCircos_data_plot_gene <- data.frame(
chromosome = ifelse(startsWith(c(data_sv_plot[[Site1_Chromosome]],
data_sv_plot[[Site2_Chromosome]]), "chr"),
c(data_sv_plot[[Site1_Chromosome]],
data_sv_plot[[Site2_Chromosome]]),
paste0(c("chr"), c(data_sv_plot[[Site1_Chromosome]],
data_sv_plot[[Site2_Chromosome]]))
),
chromStart = c(data_sv_plot[[Site1_Position]],
data_sv_plot[[Site2_Position]]),
chromEnd = c(data_sv_plot[[Site1_Position]],
data_sv_plot[[Site2_Position]]),
Gene = c(data_sv_plot[[Site1_Hugo_Symbol]],
data_sv_plot[[Site2_Hugo_Symbol]])
)
RCircos_data_plot_link <- data.frame(
chromosome = ifelse(startsWith(data_sv_plot[[Site1_Chromosome]], "chr"),
c(data_sv_plot[[Site1_Chromosome]]),
paste0(c("chr"), data_sv_plot[[Site1_Chromosome]])),
chromStart = as.numeric(data_sv_plot[[Site1_Position]]),
chromEnd = as.numeric(data_sv_plot[[Site1_Position]]),
chromosome.1 = ifelse(startsWith(data_sv_plot[[Site2_Chromosome]], "chr"),
c(data_sv_plot[[Site2_Chromosome]]),
paste0(c("chr"), data_sv_plot[[Site2_Chromosome]])),
chromStart.1 = as.numeric(data_sv_plot[[Site2_Position]]),
chromEnd.1 = as.numeric(data_sv_plot[[Site2_Position]]))
library(RCircos)
if (genome=="hg19") {
data(UCSC.HG19.Human.CytoBandIdeogram)
RCiros.cyto <- UCSC.HG19.Human.CytoBandIdeogram
}
if (genome=="hg38") {
data(UCSC.HG38.Human.CytoBandIdeogram)
RCiros.cyto <- UCSC.HG38.Human.CytoBandIdeogram
}
if (label){track_nubmer=3} else{track_nubmer=2}
RCircos.Set.Core.Components(cyto.info=RCiros.cyto, chr.exclude=NULL,
tracks.inside=track_nubmer, tracks.outside=0)
RCircos.Set.Plot.Area()
title(title)
# Draw chromosome ideogram
RCircos.Chromosome.Ideogram.Plot()
# ## 为了图的美观,不显示所有的基因
rcircos.params <- RCircos.Get.Plot.Parameters()
unlist(rcircos.params)
# # rcircos.params$base.per.unit=30000
# rcircos.params$char.width = 500
# rcircos.params$text.size = 0.4
# RCircos.Reset.Plot.Parameters(rcircos.params)
# #the maxLabels are updated accordingly
# RCircos.Get.Gene.Name.Plot.Parameters()
############################# 标注基因位置,第1个track放置连线,第2个track放基因名文字
# Connectors in first track and gene names in the second track.
# data(RCircos.Gene.Label.Data);
# > RCircos.Gene.Label.Data
# chromosome chromStart chromEnd Gene
# 1 chr1 8921418 8934967 ENO1
# Connectors in first track and gene names in the second track.
if (label){
RCircos_data_plot_gene$chromStart = as.numeric(RCircos_data_plot_gene$chromStart)
RCircos_data_plot_gene$chromEnd = as.numeric(RCircos_data_plot_gene$chromEnd)
RCircos_data_plot_gene <- RCircos_data_plot_gene[!(duplicated(RCircos_data_plot_gene[,c("chromosome", "Gene")])),]
RCircos.Gene.Connector.Plot(genomic.data=RCircos_data_plot_gene,
track.num=1, side="in")
RCircos.Gene.Name.Plot(gene.data=RCircos_data_plot_gene, name.col=4,
track.num=2, side="in")
}
############################# fusion link
# data(RCircos.Link.Data)
# # > RCircos.Link.Data
# # chromosome chromStart chromEnd chromosome.1 chromStart.1 chromEnd.1
# # 1 chr1 8284703 8285399 chr1 8285752 8286389
if (label){track_nubmer_link=3} else{track_nubmer_link=1}
# RCircos.Link.Plot(link.data=RCircos_data_plot_link, track.num=track_nubmer_link,
# by.chromosome=color_by_chr)
RCircos.Get.Link.Colors(RCircos_data_plot_link, genomic.columns=3, by.chromosome=color_by_chr)
RCircos_data_plot_link["PlotColor"] <- RCircos.Get.Link.Colors(RCircos_data_plot_link,
genomic.columns=3,
by.chromosome=color_by_chr)
if (color_by_chr){
RCircos_data_plot_link["PlotColor"] <- ifelse(RCircos_data_plot_link[["PlotColor"]] == "#FF000040", col_red,
ifelse(RCircos_data_plot_link[["PlotColor"]] == "#0000FF40", col_blue,
RCircos_data_plot_link[["PlotColor"]]))
# print(RCircos_data_plot_link["PlotColor"])
}
# RCircos_data_plot_link <- RCircos_data_plot_link[order(data_sv_plot$Frequency,decreasing=TRUE),]
#
if (length(color_col) >0){
RCircos_data_plot_link["PlotColor"] <- data_sv_plot[[color_col]]
}
RCircos.Link.Plot(link.data=RCircos_data_plot_link,
track.num=track_nubmer_link,
by.chromosome=FALSE)
data_sv_plot$plot_color <- RCircos_data_plot_link["PlotColor"]
return(data_sv_plot)
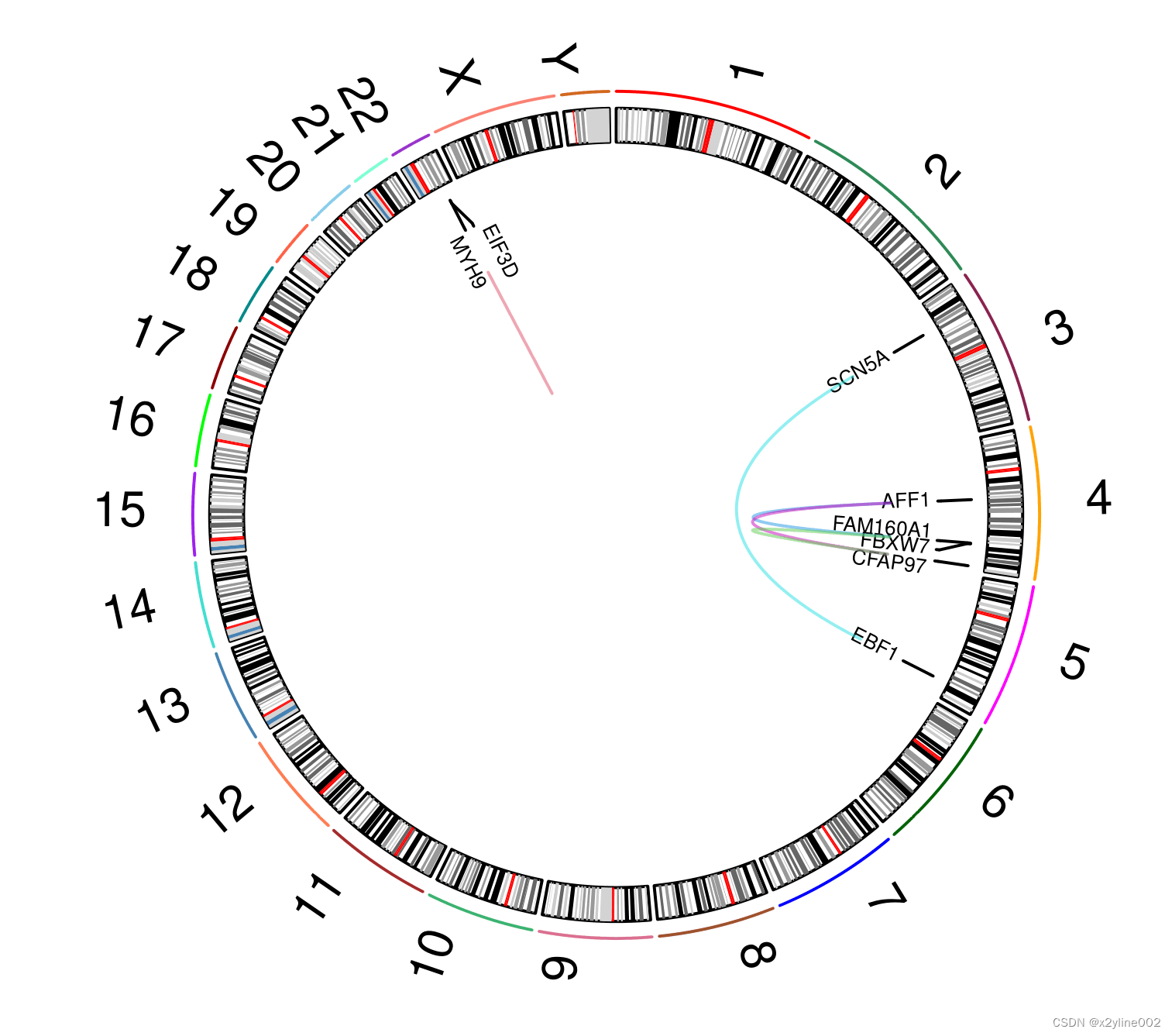
}最后输出的pdf绘图效果:















 4399
4399

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









