







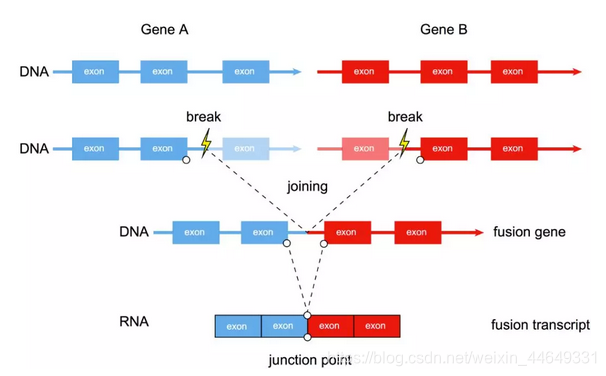
融合基因介绍
概念
在RNA水平上,由多个转录本构成的转录本。
在DNA水平上,由两个或多个基因共同组成的新基因。
NGS如何鉴定融合基因
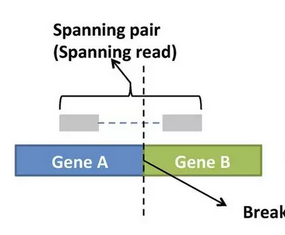
- spanning reads
R1和R2没有覆盖到连接点,只是比对的位置位于两个不同的基因上。潜在的融合基因,解释性较弱。
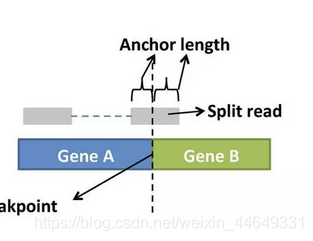
- split reads
R1或R2的一条read位于连接点的两侧,有一条read直接覆盖到连接点上。解释性较强。
本质
染色体重排。
研究意义
异常基因融合可能引用恶性血液疾病以及肿瘤。探讨发病机制、biomaker的筛选。
STAR-Fusion
原理
- 将reads通过STAR比对reference genome,筛选出split和dicordant reads作为候选融合基因。
- 候选融合基因与reference genome比对,根据ove
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章


















 302
302

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









