






Variant caller
文章目录
汇总
列举软件和文献
可检测SNV/InDel软件:
- MutScan:先构建变异信息库(上下游30bp),然后利用kmer图编辑距离从输入的fq和信息库进行变异检测;【输入fastq】
- LoFreq:考虑测序过程中的错误和偏差,通过局部重比对和伯努利实验构建测序错误模型; 【输入BAM】
- Strelka2:采用层次化的等位基因模型、混合模型方法估计插入/删除误差、正常样本污染模型以及随机森林模型进行实证变体再评分等多种先进的算法和技术;【输入BAM】
- FreeBayes: 基于贝叶斯单倍型的,考虑测序数据的复杂性,如测序深度、碱基质量和比对质量等;【输入BAM】
- VarDict:基于重比对检测SNV/Indel/大片段插入缺失SV/复杂变异;【输入BAM】
- SomaticSniper:通过比较肿瘤(T)和正常(N)样本的基因型(G)数据,计算每个位点的体细胞评分(S);【输入BAM】
- Pindel:基于一种寻找最小和最大uniq序列的搜索方式(pattener grouth),对大片段缺失和中片段插入进行检测;【输入BAM】
Scalpel:基于滑动窗口和配对末端测序(PE)数据的算法。【网上搜不到文献支持】
| 序号 | Software | 软件地址 | 文献地址 | 发表时间 |
|---|---|---|---|---|
| 1 | MutScan [ 1 ] ^{[1]} [1] | http://www.github.com/OpenGene/MutScan | https://doi.org/10.1186/s12859-018-2024-6 | 2018 |
| 2 | LoFreq [ 2 ] ^{[2]} [2] | https://csb5.github.io/lofreq/ https://github.com/andreas-wilm/lofreq3 | https://doi.org/10.1093/nar/gks918 | 2012 |
| 3 | Strelka2 [ 3 ] ^{[3]} [3] | https://github.com/Illumina/strelka | https://doi.org/10.1038/s41592-018-0051-x | 2018 |
| 4 | FreeBayes [ 4 ] ^{[4]} [4] | https://github.com/freebayes/freebayes | https://arxiv.org/abs/1207.3907 | 2012 |
| 5 | VarDict [ 5 ] ^{[5]} [5] | https://github.com/AstraZeneca-NGS/VarDict | https://doi.org/10.1093/nar/gkw227 | 2016 |
| 6 | SomaticSniper [ 6 ] ^{[6]} [6] | https://github.com/genome/somatic-sniper | https://doi.org/10.1093/bioinformatics/btr665 | 2011 |
| 7 | Pindel [ 7 ] ^{[7]} [7] | https://github.com/genome/pindel | https://doi.org/10.1093/bioinformatics/btp394 | 2009 |
参考文献:
- [1] Chen, S., Huang, T., Wen, T. et al. MutScan: fast detection and visualization of target mutations by scanning FASTQ data. BMC Bioinformatics 19, 16 (2018).
- [2] Wilm A, Aw PP, Bertrand D, Yeo GH, Ong SH, Wong CH, Khor CC, Petric R, Hibberd ML, Nagarajan N. LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. 2012 Dec;40(22):11189-201. doi: 10.1093/nar/gks918.
- [3] Kim, S., Scheffler, K., Halpern, A.L. et al. Strelka2: fast and accurate calling of germline and somatic variants. Nat Methods 15, 591–594 (2018). https://doi.org/10.1038/s41592-018-0051-x
- [4] Garrison EP, Marth GT. 2012. Haplotype-based variant detection from short-read sequencing. arXiv:1207.3907.
- [5] Lai Z, Markovets A, Ahdesmaki M, Chapman B, Hofmann O, McEwen R, Johnson J, Dougherty B, Barrett JC, and Dry JR. VarDict: a novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res. 2016, pii: gkw227.
- [6] Larson, David E., Harris, Christopher C., Chen, Ken., Koboldt, Daniel C., & Abbott, Travis E… (2011). SomaticSniper: identification of somatic point mutations in whole genome sequencing data. Bioinformatics (Oxford, England), 28(3), 311-7.
- [7] Ye, Kai., Schulz, Marcel H., Long, Quan., Apweiler, Rolf., & Ning, Zemin… (2009). Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics (Oxford, England), 25(21), 2865-71.
可检测SV软件:
- Aperture:采用k-mer依赖的方法在3个不同的库中进行快速断点查找。断点快速检测采用二进制标签的方法。最终输出结果包括断点位置、分子数、比对质量值等信息。
- BreakDancer:基于配对末端测序(PE)数据的算法,提供了详细的变异信息和可视化工具。
待更新
软件文献下载:
链接: https://pan.baidu.com/s/1NAdMYgjh-AHn8-lZ0U1Zbg 提取码: mi9n
软件比较文献
- EBCall
- Mutect
- Seurat
- Shimmer
- Indelocator
- SomaticSniper
- Strelka
- VarScan 2
- Virmid
2018年文献几十款找somatic mutation的软件: A review of somatic single nucleotide variant calling algorithms fornext-generation sequencing data
- BAYSIC
- CaVEMan
- deepSNV
- EBCall
- FaSD-somatic
- FreeByes
- HapMuC
- JointSNVMix2
- LocHap
- LoFreq
- LoLoPicker
- MutationSeq
- MuSE
- MuTect
- SAMTools
- Platypus
- qSNP
- RADIA
- Seurat
- Shimmer
- SNooPer
- SNVSniffer
- SOATsnv
- SomaticSeq
- SomaticSniper
- Strelka
- TVC
- VarDict
- VarScan2
- Virmid
TCGA计划软件
公众号参考:http://www.bio-info-trainee.com/7702.html、https://mp.weixin.qq.com/s/BXSjZsK4Jf-Tu1jDpA6YNg
TCGA计划采取的体细胞突变(somatic mutation)检测软件:
- MuSE
- VarScan
- MuTect
- SomaticSniper
Scalable Open Science Approach for Mutation Calling of Tumor Exomes Using Multiple Genomic Pipelines【March-2018/10.1016/j.cels.2018.03.002】
| Software and Algorithms | GitHub |
|---|---|
| MuTect | https://github.com/broadinstitute/mutect |
| Pindel | https://github.com/genome/pindel |
| Radia | https://github.com/aradenbaugh/radia |
| VarScan2 | http://dkoboldt.github.io/varscan/ |
| SomaticSniper | https://github.com/genome/somatic-sniper |
| MuSE | https://github.com/danielfan/MuSE |
| Indelocator | http://archive.broadinstitute.org/cancer/cga/indelocator |
| Maf2Vcf | https://github.com/covingto/vcf2maf/ |
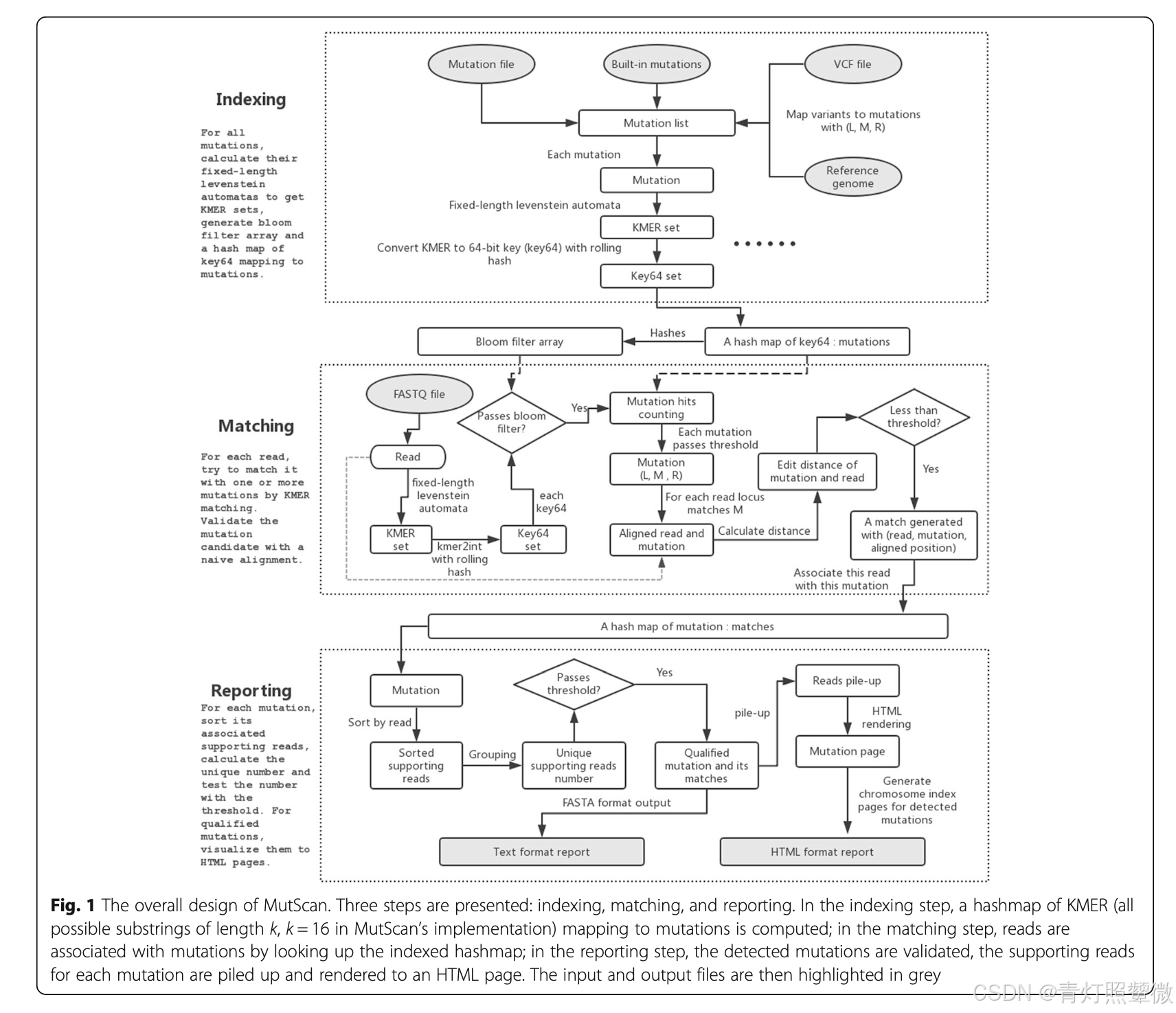
MutScan
Chen, S., Huang, T., Wen, T. et al. MutScan: fast detection and visualization of target mutations by scanning FASTQ data. BMC Bioinformatics 19, 16 (2018).
-
功能:直接扫描FastQ文件以检测目标变异,特别适用于癌症相关的药物靶点检测。
-
特点:无需预先排列和变异调用,敏感度高,运行速度快,提供美观的HTML报告。
-
软件地址:http://www.github.com/OpenGene/MutScan
-
软件文献:Chen, S., Huang, T., Wen, T. et al. MutScan: fast detection and visualization of target mutations by scanning FASTQ data. BMC Bioinformatics 19, 16 (2018). https://doi.org/10.1186/s12859-018-2024-6
-
使用示例:
mutscan -1 <read1_file_name> -2 <read2_file_name> -h report.html mutscan -1 R1.fq -2 R2.fq -m target.vcf -r hg19.fa # --mutation 默认是有60个与癌症相关的位点库 mutscan --read1 $r1 --read2 $r2 --mutation $mutDB -h $html > $outvar
软件流程图:
LoFreq
Wilm A, Aw PP, Bertrand D, Yeo GH, Ong SH, Wong CH, Khor CC, Petric R, Hibberd ML, Nagarajan N. LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. 2012 Dec;40(22):11189-201. doi: 10.1093/nar/gks918.
-
功能:一款专注于低频变异的检测软件,适用于癌症和遗传病研究。
-
特点:能够处理低覆盖度和低质量的测序数据,提高低频变异的检测准确性。
-
软件网址: https://csb5.github.io/lofreq/、https://github.com/andreas-wilm/lofreq3
-
文献网址:https://doi.org/10.1093/nar/gks918
-
使用示例:输入BAM
lofreq call -f ref.fa -o vars.vcf aln.bam lofreq call-parallel --pp-threads 8 -f ref.fa -o vars.vcf aln.bam lofreq somatic -n normal.bam -t tumor.bam -f hg19.fa --threads 8 -o out_ [-d dbsnp.vcf.gz]
LoFreq在处理数据时,会考虑多种因素,如测序深度、碱基质量分数、局部序列上下文等,以提高变异检测的准确性和灵敏度。它还会使用特定的算法来过滤掉可能的假阳性结果,如测序错误或正常样本中的背景变异。具体的,LoFreq会先基于伯努利实验(Bernoulli trial)构建测序错误模型,并对计算时间进行优化。在没有用户提供的质量值的情况下,LoFreq对所有12个可能的基替换类的错误率进行建模,并使用预期最大化(EM)方法来估计。
Strelka2
Kim, S., Scheffler, K., Halpern, A.L. et al. Strelka2: fast and accurate calling of germline and somatic variants. Nat Methods 15, 591–594 (2018).
-
功能:另一款用于检测体细胞变异的软件。
-
特点:与Mutect2类似,但具有不同的算法和参数设置,适用于不同的数据集和分析需求。
-
软件地址:https://github.com/Illumina/strelka
-
文献地址:https://doi.org/10.1038/s41592-018-0051-x
-
使用示例:
strelka/strelka-2.8.2.centos5_x86_64/bin/configureStrelkaSomaticWorkflow.py \ --normalBam $normal_bam \ --tumorBam $tumor_bam \ --referenceFasta $reference \ --runDir somatic/strelka
FreeBayes
Garrison E, Marth G. Haplotype-based variant detection from short-read sequencing. arXiv preprint arXiv:1207.3907 [q-bio.GN] 2012
-
功能:一款基于贝叶斯单倍型的变异检测软件,适用于多种类型的变异检测,包括SNP、INDEL等。
-
特点:在植物变异检测中表现出色,效率高且准确性好。同时,FreeBayes也支持多线程并行计算,能够处理大规模的基因组数据。考虑了测序数据的复杂性,如测序深度、碱基质量和比对质量等。
-
软件地址:https://github.com/freebayes/freebayes
-
文献地址:https://arxiv.org/abs/1207.3907
-
使用示例:
freebayes -f ref.fa aln.cram >var.vcf # 覆盖深度要求 freebayes -f ref.fa aln.bam --gvcf -g 1000 >var.vcf # 特定位置区域 freebayes -f ref.fa -r chrQ aln.bam >var.vcf freebayes -f ref.fa -r chrQ:1000-2000 aln.bam >var.vcf # 至少有5条支持 freebayes -f ref.fa -C 5 aln.bam >var.vcf # 更多示例见软件地址 ...
软件作者的博客介绍freebayes:http://ekg.github.io/2015/12/08/How-to-freebayes
VarDict
Lai Z, Markovets A, Ahdesmaki M, Chapman B, Hofmann O, McEwen R, Johnson J, Dougherty B, Barrett JC, and Dry JR. VarDict: a novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res. 2016, pii: gkw227.
求臻 打断测序错误文献中提到过VarDict:2024.03-04.4-H.Chen-Characterization and mitigation of artifacts derived from NGS library preparation due to structure-specific sequences in the human genome
-
功能:一款基于Python的变异检测工具,能够识别单核苷酸多态性(SNP)、插入/删除(INDEL)以及结构变异(SV)等。
-
特点:采用了基于哈希表的算法,支持多种变异类型(如SNP、INDEL等)的检测,并且具有较高的准确性和灵敏度。
-
软件网址:https://github.com/AstraZeneca-NGS/VarDict
-
文献网址:https://doi.org/10.1093/nar/gkw227
-
使用示例:输入BAM
# 单个样本 AF_THR="0.01" # minimum allele frequency vardict -G /path/to/hg19.fa -f $AF_THR -N sample_name -b /path/to/my.bam -c 1 -S 2 -E 3 -g 4 /path/to/my.bed | teststrandbias.R | var2vcf_valid.pl -N sample_name -E -f $AF_THR # Tumor Normal配对样本 AF_THR="0.01" # minimum allele frequency vardict -G /path/to/hg19.fa -f $AF_THR -N tumor_sample_name -b "/path/to/tumor.bam|/path/to/normal.bam" -c 1 -S 2 -E 3 -g 4 /path/to/my.bed | testsomatic.R | var2vcf_paired.pl -N "tumor_sample_name|normal_sample_name" -f $AF_THR
VarDict可同时检测SNV、MNV、InDels、复杂和结构变体。【识别潜在位点】对测序结果进行解读,识别出潜在的变异位点;【Fisher’s精确检验】在配对样本分析模式下,VarDict会提取变异和参考等位基因的reads数,并使用Fisher’s精确检验来确定变异在两个样本之间是否存在显著差异,从而分类为体细胞/胚系变异/LOH(杂合性缺失);【局部重比对】对于复杂的变异,如大InDel和结构变异,VarDict采用局部重比对策略,通过比对测序读取与参考基因组之间的局部序列差异,来准确识别这些变异。
另外,在基于扩增子变异检测,能够识别PCR错误问题。软件文献中所述的PCR错误:
-
错误特征:(1)两个有重叠扩增子,一个有变异,另一个无。(2)引物相似:比如左端是ERBB2引物(相似引物)、右端是EGFR(7号)引物。
-
错误原因:PCR错误、引物相似问题。
-
解决方法:识别后直接去除。
VUS (variant of unknown significance)
SomaticSniper
Larson, David E., Harris, Christopher C., Chen, Ken., Koboldt, Daniel C., & Abbott, Travis E… (2011). SomaticSniper: identification of somatic point mutations in whole genome sequencing data. Bioinformatics (Oxford, England), 28(3), 311-7.
-
功能:专门用于检测体细胞变异的软件,适用于癌症基因组学研究。
-
特点:通过比较肿瘤(T)和正常(N)样本的基因型(G)数据,计算每个位点的体细胞评分(S)。能够处理肿瘤和正常样本的配对数据,提高体细胞变异的检测准确性。
-
软件地址:https://github.com/genome/somatic-sniper
-
文献地址:https://doi.org/10.1093/bioinformatics/btr665
-
使用示例:(使用的人可能不多,网搜相关信息较少,作者还发表了一篇使用软件文献:Using SomaticSniper to Detect Somatic Single Nucleotide Variants)
bam-somaticsniper –f ref.fa tumor.bam normal.bam output.txt
文献中提到与软件SNVMix2进行比较,灵敏度略高:
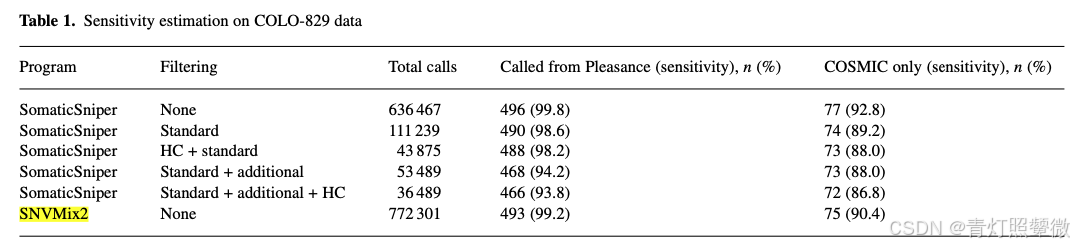
虽然网搜SomaticSniper相关信息不多,但是文献中评分规则可以参考:
(1)计算某一位点非体细胞突变的似然度,通过比较肿瘤和正常样本在该位点的基因型似然度来评估。
(2)使用MAQ算法计算基因型似然度。
(3)根据不同情况(如杂合子与参考碱基共享等位基因、纯合子变异等)计算先验概率。
(4)通常设定体细胞评分阈值为15,低于此阈值的位点通常不被报告为体细胞突变。



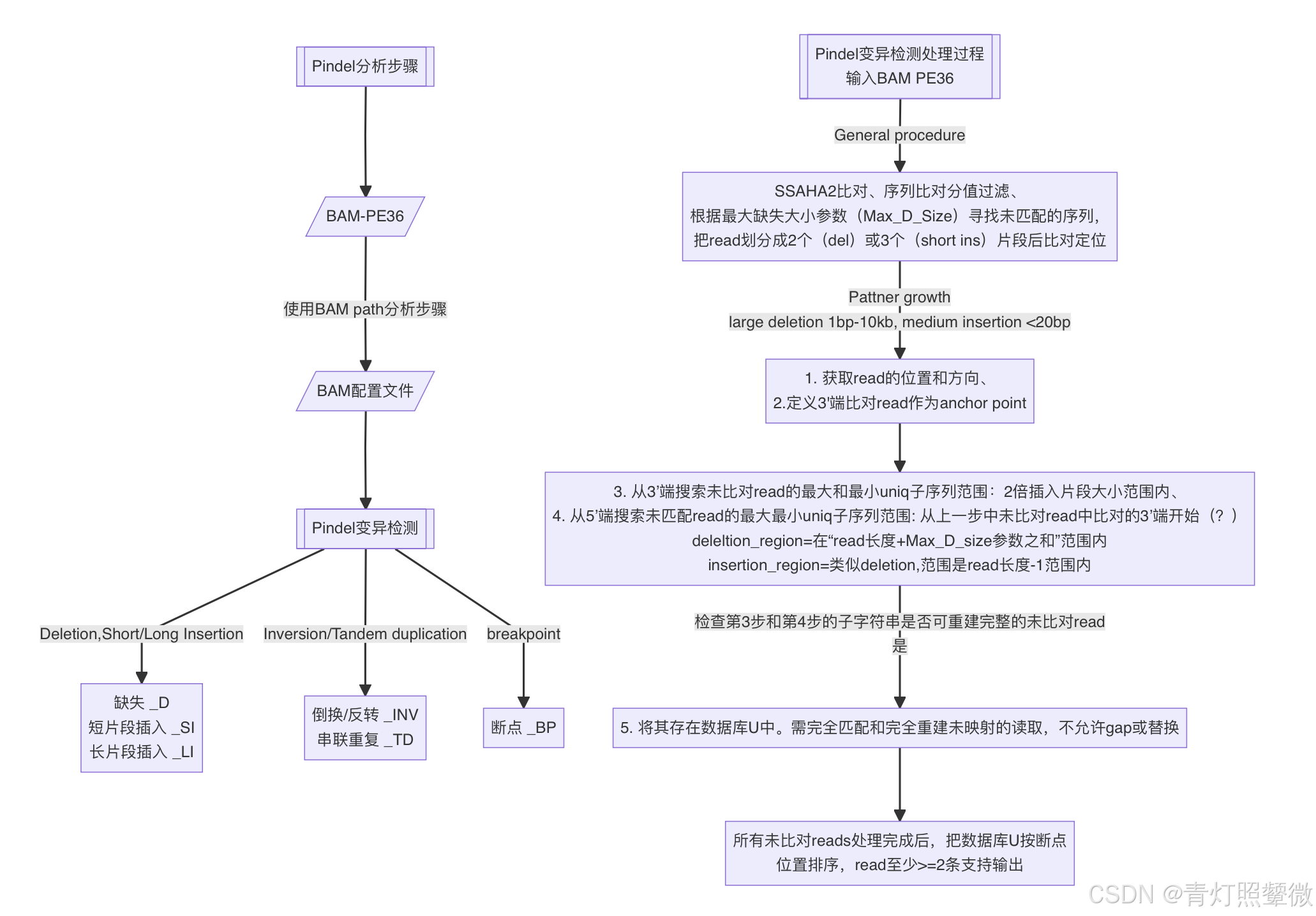
Pindel
Ye, Kai., Schulz, Marcel H., Long, Quan., Apweiler, Rolf., & Ning, Zemin… (2009). Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics (Oxford, England), 25(21), 2865-71.
-
功能:检测插入(<=20)和删除(1bp-10kb)变异,特别适用于检测较大的DEL变异。
-
特点:具有较高的准确性和灵敏度。采用了划分read并使用最大最小uniq子序列的算法,能够准确检测INDEL和SV。它提供了灵活的参数设置和多种输出格式,以满足不同用户的需求。
-
软件地址:https://github.com/genome/pindel
-
文献地址:https://doi.org/10.1093/bioinformatics/btp394
-
使用示例:参考软件地址FAQ和http://gmt.genome.wustl.edu/packages/pindel/quick-start.html
./pindel -f <reference.fa> -p <pindel_input> [and/or -i bam_configuration_file] -c <chromosome_name> -o <prefix_for_output_files> # 示例1: ./pindel -f test/SmallTest/sim1chrVs2.fa -i test/SmallTest/sim1chrVs2.conf_for_demo -o simulated_test.out # 示例2: ./pindel -f demo/hs_ref_chr20.fa -p demo/COLO-829_20-p_ok.txt -c 20 -o output/ref ./pindel -f demo/simulated_reference.fa -i demo/simulated_config.txt -c ALL -o output/simulated参考博客:https://www.cnblogs.com/lmt921108/p/13964733.html
Pindel变异检测过程:文章数据使用双端PE36的测序数据
samtools mpilup
- 功能:生成测序数据的pileup格式文件,用于后续变异检测分析。
- 特点:作为samtools软件包的一部分,提供高效、准确的pileup文件生成功能。Joint genotype analysis
# 1. 生成mpileup文件
samtools mpileup -f reference.fa input.sorted.bam > output.pileup
# 2. bcftools检变异
bcftools call -mv output.pileup > output.raw.vcf
# 3. 变异过滤
bcftools filter -sLowQual -e'%QUAL<20 || DP>100' output.raw.vcf > output.flt.vcf
GATK Mutect2
- 功能:专门用于检测体细胞变异的软件,适用于癌症研究。
- 特点:能够处理复杂的肿瘤样本数据,提高体细胞变异的检测准确性。
GATK HaplotypeCaller:
- 功能:GATK(Genome Analysis Toolkit)中的变异检测模块,能够检测SNP、INDEL等类型的变异。
- 特点:准确性高,适用于大规模基因组数据的分析。
sentieon
- 功能:提供完整的基因变异检测二级分析方案,大幅提升测序数据的分析效率和检出精度。
- 特点:金标准数学模型,适配多种CPU计算架构,可灵活部署,被多个高影响力刊物引用。
UVC
该算法旨在精确检测germline(胚系)和somatic(体细胞)的微小突变。
- 功能:专注于变异检测,能够精准地检测somatic变异,包括SNVs和IndDels。
- 特点:在多个数据集上表现优异,产生变异质量值参数,适用于UMI标签和TN配对数据检测,运行速度快。
VarScan
- 功能:对大规模平行测序数据进行变异检测,能够检测SNP和INDEL。
- 特点:开源且兼容多种比对软件,高敏感性和特异度,提供多个参数供用户设置。
DeepVariant
- 功能:利用深度学习技术提高变异检测的准确性。
- 特点:支持多种测序技术,易于使用,支持Docker等容器化技术。
融合检测
-
DNA:BreakDancer、CREST
-
RNA:FusionMap、SoapFuse、STAR-fusion、tophat-fusion
BreakDancer
- 功能:专门用于检测结构变异,如染色体断裂、易位等。
- 特点:采用了基于配对末端测序(PE)数据的算法,能够准确识别结构变异。它提供了详细的变异信息和可视化工具,方便用户进行进一步的分析和验证。
CREST
-
功能:主要用于识别和分析基因组中的基因融合事件,为癌症等疾病的分子诊断提供关键信息。(基于Split Read原理)
-
特点:具有高灵敏度、高特异性和准确性,能够高效地从大规模测序数据中检测出基因融合事件。
-
软件地址:https://github.com/youngmook/CREST
Aperture
- 功能:一种快速的SV(结构变异)检测软件,它利用k-mer进行搜索、二进制label进行断点检测及断点聚类、利用UMI进行consensus,从而实现对ctDNA中结构变异和病毒整合的精确检测。
- 特点:输入为原始的FASTQ文件,采用k-mer依赖的方法在3个不同的库中进行快速断点查找。断点快速检测采用二进制标签的方法。最终输出结果包括断点位置、分子数、比对质量值等信息。相对于其他SV软件具有较高的灵敏度和特异性,特别是在跨越断点的重复区域。
其他
目前没有用过也没听过:SyRi、Platypus、LUMPY、RADAR、Sniffles、MosaicSuite、SVIM、SomaticSniper、SomaticSeq、CNVkit
SyRi
- 功能:识别基因组中的结构同源和重排事件。
- 特点:提供了多种参数和选项,适用于不同的数据集和分析需求。
Platypus
- 功能:适用于多种变异检测场景的软件。
- 特点:在某些特定情况下可能具有优势,如处理复杂基因组结构或低质量测序数据时。
LUMPY
- 功能:基于比对结果的结构变异检测。
- 特点:支持多线程并行计算,提高了分析效率。
RADAR
- 功能:用于检测重复序列中的变异。
- 特点:特别适用于检测重复序列中的SNP和INDEL变异。
Sniffles
- 功能:识别包括插入、删除、倒位和易位等在内的多种结构变异。
- 特点:具有较高的准确性和灵敏度。
MosaicSuite
- 功能:专注于检测嵌合体变异。
- 特点:能够处理复杂的嵌合体变异情况,提高变异检测的准确性和灵敏度。
SVIM
- 功能:基于序列比对结果的结构变异检测。
- 特点:能够识别多种类型的结构变异,适用于大规模基因组数据的分析。
SomaticSeq
- 功能:一个集成多种体细胞变异检测工具的框架。
- 特点:用户可以根据需要选择多个体细胞变异检测工具进行集成分析,提高变异检测的准确性和可靠性。
CNVkit
- 功能:用于检测拷贝数变异(CNV)的软件。
- 特点:特别适用于检测基因组中的拷贝数扩增和缺失事件。
其他相关文献
| 文献名 | 说明 |
|---|---|
| Standard operating procedure for somatic variant refinement of sequencing data with paired tumor and normal samples | IGVcheck标准,并给出20+示例IGV说明识别变异的真假。包括正常的胚系突变、单链reads假阳、支持reads上有错配、比对质量低、碱基重复区等。 |
| Yang, Xiaofei., Zheng, Gaoyang… (2024). Pindel-TD: A Tandem Duplication Detector Based on A Pattern Growth Approach. Genomics, proteomics & bioinformatics. | Pindel新文献:https://github.com/xjtu-omics/pindel |
修改日志
- 2024.10.16:初版
- 2024.10.17:更新一些软件的详细说明

















 7769
7769

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









