









抽象
动机:高-产量未来-新一代测序技术使基因组和转录组的日益快捷,实惠测序,具有广阔的应用范围。测序数据的质量对于所有应用都至关重要。产生的数据中很大一部分包含错误,因此需要更有效的错误纠正程序。
结果:我们提出了RACER(读取错误的快速,准确校正),这是一种用于校正测序数据错误的新软件程序。RACER比现有程序具有更好的纠错性能,速度更快,所需的内存更少。为了支持我们的主张,我们在各种真实数据集上与现有的领先程序进行了广泛的比较。
可用性: RACER可从www.csd.uwo.ca/~ilie/RACER/免费用于非商业用途。
联络人:ilie@csd.uwo.ca
1引言
自动Sanger测序方法(Sanger 等,1977)通过揭示DNA分子的序列,最突出的是人类基因组的序列,彻底改变了生物学研究。该方法的局限性导致需要改进的测序技术。巨大的需求导致发现了几种所谓的下一代测序(NGS)技术,例如Illumina / Solexa,Roche / 454,Life / APG的SOLiD,Helicos BioSciences的HeliScope,Pacific Biosciences和Life的Ion Torrent;Metzker(2010)的调查提供了详细说明。
这些高通量技术以降低的成本生成大量数据,从而使应用程序的数量不断增加,包括从头进行基因组组装,基因组重测序,癌症突变发现,宏基因组学,DNA-蛋白质相互作用发现等。已经为雄心勃勃的项目打开了道路,例如1000个基因组计划(Siva,2008年),第一个对大量人的基因组进行测序以提供有关人类遗传变异的综合资源的项目,以及Genome 10K项目(Haussler 等,2009),旨在发现10000种脊椎动物的基因组。
大量的生物信息学程序对于分析NGS数据至关重要。尽管它们的多样性令人印象深刻,但它们都需要高质量的数据。当前占主导地位的技术Illumina产生的读取中有近一半包含错误。除其他问题外,它们使基因组组装更加困难,而基因组组装者,例如Euler-SR(Chaisson和Pevzner,2008),ALLPATHS(Butler 等人,2008),ABySS(Simpson 等人,2009),SOAPdenovo (Li et al。,2010)或SGA(Simpson and Durbin,2012),都包含自己的校正机制。
问题的重要性导致设计了许多独立的纠错程序,例如Euler(Chaisson 等人,2004),SHREC(Schröder 等人,2009),Reptile(Yang 等人,2010)。 ,Quake(Kelley 等,2010),CUDA(Shi 等,2010),HSHREC(Salmela,2010),SOAP(Li 等,2010),HiTEC(Ilie 等,2011),Coral(等)Salmela andSchröder,2011),Hammer(Medvedev et al。,2011)),ECHO(Kao 等,2011),PSAEC(Zhao 等,2011b)和MyHybrid(Zhao 等,2011a);另请参见Yang 等人的调查。(2013)。
测序数据中存在的错误包括:替换(错误的碱基替换为正确的碱基)和indel(插入新碱基或删除现有碱基的碱基)。但是,由于主要产生替换错误的Illumina技术的支配,大多数现有程序都将重点放在纠正此类错误上。出于同样的原因,我们新程序的当前版本也将重点放在替换错误上。
所有校正程序的主要思想基本相同。高通量技术通过高覆盖率来补偿较短的阅读长度,这意味着基因组中的每个位置均被测序了多次,其中许多是正确的。因此,在仔细的分析中,正确的方法将占上风,并且可以纠正错误。正是这种进行分析的方式使各种程序之间有所不同。
一些程序对k- mers 的数量进行计数,而那些计数超过给定阈值的程序则被认为是正确的,而其余程序则被校正。其中包括SHREC,HiTEC,HSHREC和PSAEC。其他人则认为在每个读数中都出现了所有k聚体,即k 谱(Pevzner 等,2001),并尝试以最小的编辑距离校正整个读数,以使所有k聚体的计数都高于阈值。CUDA,Quake,Reptile和Hammer包括在此处。最后,Coral,ECHO和MyHybrid基于多个序列比对。读者可参考Yang 等人的调查。(2013)。
我们提出了一个新程序,即RACER(读取中的错误的快速而准确的校正),它属于第一类。尽管SHREC和HSHREC使用后缀树,而HiTEC使用更有效的后缀数组,但是RACER使用完全不同,更有效的数据结构。这样,尽管SHREC和较小的HiTEC都具有很高的空间消耗,但是RACER的空间效率很高。与现有程序相比,它在纠正错误方面也更快,更有效。我们已经与各种真实数据集上的领先程序进行了广泛的比较。RACER可从www.csd.uwo.ca/~ilie/RACER/免费获取以用于非商业用途。
2结果
2.1数据集
我们对具有不同读取长度,基因组大小和覆盖范围的各种数据集进行了广泛的测试。仅根据真实数据集进行比较,因为根据我们的经验,模拟数据集不能很好地表明实际性能。此外,它们易于纠正,并且纠正程序经常表现出不切实际的高性能。
十五真实数据集被认为是,其中一些先前已经使用,也可以用于校正目的或组件:È scherichia大肠杆菌36,大肠杆菌47(以下名字的数字表示读取长度和用于区分它们),金黄色葡萄球菌,啤酒糖酵母和果蝇果蝇已经在调查中使用杨等人。(2013),秀丽隐杆线虫被Simpson和Durbin(2012)用来比较SGA和其他基因组装配体。。其余的数据集是新的。表1给出了数据集的登录号和详细信息,以及与参考基因组有关的相应信息。补充表S4中提供了完整的生物名称。
用于评估的数据集,越来越多地按碱基对总数排序
| 数据集 | 登记号 | 参考基因组 | 基因组长度 | 读取长度 | 读取次数 | 碱基对数 | 覆盖范围 |
|---|---|---|---|---|---|---|---|
| 乳酸乳球菌 | SRR088759 | NC_013656.1 | 2598144 | 36 | 4370050 | 157321800 | 60.55 |
| 梅毒螺旋体 | SRR361468 | CP002376.1 | 1139417 | 35 | 7133663 | 249678205 | 219.13 |
| 大肠杆菌75a | SRR396536 | NC_000913.2 | 4639675 | 75 | 3454048 | 259053600 | 55.83 |
| 枯草芽孢杆菌 | DRR000852 | NC_000964.3 | 4215606 | 75 | 3519504 | 263962800 | 62.62 |
| 大肠杆菌75b | SRR396532 | NC_000913.2 | 4639675 | 75 | 4341061 | 325579575 | 70.17 |
| 铜绿假单胞菌 | SRR396641 | NC_002516.2 | 6264404 | 36 | 9306557 | 335 036 052 | 53.48 |
| 大肠杆菌47 | SRR022918 | NC_000913.1 | 4771872 | 47 | 14408630 | 677205610 | 141.92 |
| 钩端螺旋体 | SRR353563 | NC_004342.2 | 4338762 | 100 | 7066162 | 706 616 200 | 162.86 |
| 询问乳杆菌 | SRR397962 | NC_005823.1 | 4277185 | 100 | 7127250 | 712725 000 | 166.63 |
| 大肠杆菌36 | SRX000429 | NC_000913.1 | 4771872 | 36 | 20816448 | 749392128 | 157.04 |
| 嗜血杆菌流感 | SRR065202 | NC_000907.1 | 1830138 | 42 | 23935272 | 1005281424 | 549.29 |
| 金黄色葡萄球菌 | SRR022866 | NC_003923.1 | 2901156 | 76 | 25551716 | 1941930416 | 669.36 |
| 酿酒酵母 | SRX100885 | PRJNA128 | 12416363 | 76 | 52061664 | 3956686464 | 318.67 |
| 线虫 | SRR065390 | wormbase.org | 102291899 | 100 | 67617092 | 6761709200 | 66.10 |
| 黑腹果蝇 | SRX006151 | flybase.org | 120220296 | 45 75 95 | 101548652 | 6903898304 | 57.43 |
| SRX006152 | |||||||
| SRX023452 |
注:所有数据集和参考基因组序列是从国家除外生物技术信息中心获得线虫从www.wormbase.org和果蝇从flybase.org。
2.2评估
我们已经将RACER与Yang 等人测试过的程序进行了比较。(2013),即HiTEC,SHREC,Reptile,Quake,Coral,Coral,SOAP和ECHO。(由于仅考虑替换错误,所以我们测试了SHREC而不是HSHREC。)其中,由于空间或时间要求高,SOAP和ECHO无法运行大多数数据集,因此无法进行比较。
在表1中的原始数据集上评估了所有测试程序的纠正错误的能力以及所需的时间和空间。纠错性能评估为纠正的错误百分比。我们的评估方法具有避免映射或汇编程序干扰的优势,方法的3.2节对此进行了详细介绍。
为了进行统一比较,我们通过将时间和空间值除以数据集中碱基对的总数来对其进行归一化。实际的时间和空间值在补充材料中给出。
我们已经在同一台惠普计算机上运行了所有程序,该计算机具有2.1 GHz的24核AMD Opteron和98 GB的随机存取内存,运行Linux Red Hat,CentOS 5.5 m。由于HiTEC和Reptile不并行运行,因此我们在串行和并行模式下都进行了测试,以将所有程序都包括在比较中。我们已经在并行模式下使用了24个内核。
结果显示在表2中,其中包含在顶部的纠错性能(以纠错的百分比表示),在中间的时间(以每兆字节输入碱基对的秒为单位)和在底部的空间(以兆字节为单位)输入碱基对。某些程序无法运行所有数据集,并且原因由表标题中解释的字母指示。每个部分的最后一行代表可以运行所有程序的那些数据集的条目平均值。为了改善表格的可视化效果,我们使用了颜色强度,深色表示更好的性能。

2.3比较
就纠错性能而言,只有HiTEC才能接近RACER。但是,HiTEC需要第二高的空间,因此无法运行最大的数据集。对于D.melanogaster,由于读取长度不同,HiTEC并未尝试运行它。另请注意,HiTEC在某些未包含在平均值中的数据集上的性能较差。直接比较显示出显着差异。
在时间方面,RACER在串行和并行模式下均是最快的,比串行第二名Quake快两倍,并且在并行模式下比所有其他Quake快大约一个数量级。RACER还比其他程序更具存储效率。与并行模式下的RACER相比,Quake再次名列第二,并且使用的空间大于50%。
RACER和Reptile是唯一能够运行所有数据集的程序。Quake在时间和空间上排在第二位,串行空间除外,排在第三位,但其纠错性能在串行和并行方面均排在倒数第二,大约是RACER的一半。部分原因是由于Quake修剪无法校正时读取。评估计算所有正确读取中碱基对的数量。爬行动物在时间上排在第三位,在空间上排在第二位(串行模式),但其准确性却是倒数第二。SHREC显然需要最大的空间,因此它能够运行我们计算机上最少的数据集。另一方面,它在校正性能方面始终居第三位。在串行模式下,Coral速度最慢,但在所有程序中速度最高,并且其并行性能接近Quake和SHREC。15倍加速是以增加12倍空间为代价的。RACER的速度提高了11倍,空间增加了一点。
3种方法
3.1赛车
RACER属于k- mer计数程序的类别。它使用2位核苷酸编码和未知位置的随机替换。每个k- mer表示为一个64位整数。的ķ聚体被存储在哈希表中。对于每个k聚体,计算对应于两侧上八个可能的核苷酸的八个计数器。阈值t用于区分正确位置和错误位置。甲ķ -mer随后是核苷酸一个假设正确的,如果该计数高于吨和错误否则。在后一种情况下,假设恰好有一个字母b紧跟相同的k-mer并且计数高于t,a替换为b。
需要输入测序的基因组的近似大小作为输入,RACER根据该大小自动计算校正部分中使用的k- mer大小k和阈值t。所使用的参数组合已通过实验确定。与HiTEC一样,阈值也有所不同,以实现更高的校正性能。
避免全文索引(例如后缀树和后缀数组)可以提供比SHREC和HiTEC更大的空间优势。相同的原因也带来了速度优势,因为一旦计算了k个单体和计数器,就可以多次使用它进行校正。
RACER校正来自fasta和fastq数据的不同长度的读取。它已经在C ++和OpenMP中实现,并且不需要其他软件即可运行它。
3.2评估细节
为了获得最准确的评估,我们避免了使用绘图程序或将校正后的数据用于其他应用程序,例如基因组组装。映射程序用于评估,以了解错误在哪里以及应如何纠正。但是,许多读取不能唯一地映射,甚至根本无法映射更多。此类读取将被丢弃,因此,所获得的数据集与原始数据集明显不同。特别是,所有程序都人为地提高了性能,因为最难纠正的读取已通过映射消除。另一方面,基因组组装者具有其自身的隐式或显式校正程序,并且无法预测两种校正方法之间的相互作用。
根据是否可以通过精确搜索算法在参考基因组中分别找到,将读物分类为正确或错误。分别通过e b和e a表示校正前后错误读取中碱基对的数目,纠错性能计算为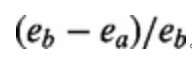 。因为我们有
。因为我们有
![]() (Ilie 等人,2011),所以我们得到
(Ilie 等人,2011),所以我们得到
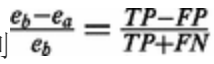
的公式与Yang 等人的公式相同。(2013年)。如上所述,通过考虑全读校正,而不是基于个体的校正,可以期望达到类似的性能;但是,避免了与读取映射相关的问题。
但是,由于在某些文章中是对映射的数据集进行比较,因此为了完整起见,我们在补充材料中提供了这种比较,其中使用BWA(Li和Durbin,2009年)以较高的错误率对数据集进行了映射,因此保持大量读取,并最大程度降低了偏差。在映射的数据集上进行的比较与在原始数据集上进行的比较相似,不同之处在于,如预期的那样,校正的错误百分比增加了。
所有程序均已按照作者在相应文章,网站或自述文件中给出的说明运行。我们没有调整任何程序的参数来提高性能,因为这是不现实的。用于运行所有程序的命令在补充材料中给出。
4。讨论
广泛的测试表明,RACER在所有方面均优于现有程序:纠错性能,时间和空间。对于细菌数据集,它在<2分钟内校正了大约四分之三的误差,对于蠕虫或蝇类等较大的有机体,它可以在30–40分钟内校正。RACER不需要任何其他软件即可运行。
5结论
我们提供了一种新的工具RACER,用于纠正NGS数据中的错误。当前版本的RACER定位于Illumina数据,从而纠正替换错误。未来的版本将能够处理插入缺失错误,从而能够更正来自其他测序平台的数据以及混合数据。
















 272
272











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











