







请一定看这里:写下来只是为了记录一些自己的实践,当然如果能对你有所帮助那就更好了,欢迎大家和我交流

三者区别
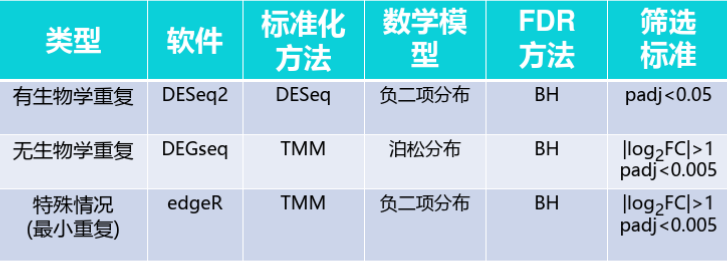
三者区别
差异分析流程:
1 初始数据
2 标准化(normalization):DESeq、TMM等
为什么要标准化?
消除文库大小不同,测序深度对差异分析结果的影响
怎样标准化?
找到一个能反映文库大小的因子,利用这个因子对rawdata进行标准化
3 根据模型检验求p value:泊松分布(poisson distribution)、负二项式分布(NB)等
4 多重假设得FDR值:
检验方法:Wald、LRT
多重检验:BH
5 差异基因筛选:pvalue、padj
💥💥💥一、 edgeR的使用💥💥💥
因为目前没有合适的数据,所以数据来源于这里 参考这篇:刘尧科学网博客
0. 前期工作
- 用到的
gene.txt文件,内容如下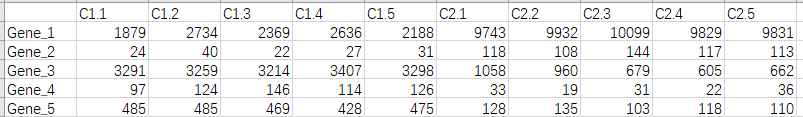
gene.txt文件内容
c1表示为一组,c2表示为另一组,.后为第几个样本
读取数据并设置分组,要保证样本名称和分组名称的顺序是一一对应的。
#加载edgeR包
library(edgeR)
#读进来文件
targets <- as.matrix(read.delim("你的路径/gene.txt", sep = '\t', row.names = 1))
#分组,这里是两组,每组5个样本
group <- rep(c('c1','c2'),each = 5)
1. 构建DGEList对象
根据基因表达量矩阵以及样本分组信息构建DGEList对象
dgelist <- DGEList(counts = targets, group = group)
2. 过滤低表达的基因
DESeq2能够自动识别这些低表达量的基因的,所以使用DESeq2时无需手动过滤。edgeR推荐根据CPM(count-per-million)值进行过滤,即原始reads count除以总reads数乘以1,000,000,使用此类计算方式时,如果不同样品之间存在某些基因的表达值极高或者极低,由于它们对细胞中分子总数的影响较大(也就是公式中的分母较大), 有可能导致标准化之后这些基因不存在表达差异,而原本没有差异的基因在标准化之后却显示出差异
这里参考这篇:当我们在说RNA-seq reads count标准化时,其实在说什么?
为了解决上述问题,BSM(between-sample normalization)类分出control set去评估测序深度而不是用所有数据,主要分三种:
(1)TMM(trimmed mean of M-values): TMM是M-值的加权截尾均值,即选定一个样品为参照,其它样品中基因的表达相对于参照样品中对应基因表达倍数的log2值定义为M-值。随后去除M-值中最高和最低的30%,剩下的M值计算加权平均值,权重来自Binomial data的delta方法 (Robinson and Oshlack, 2010)。
(2)RLE (relative log expression):首先计算每个基因在所有样品中表达的几何平均值。然后再计算该值与每个样品的比值的中位数,也叫被称为量化因子scale factor (Anders and Huber 2010)。
(3)UQ (upper quartile): 上四分位数 (upper quartile, UQ)是样品中所有基因的表达除以处于上四分位数的基因的表达值。同时为了保证表达水平的相对稳定,计算得到的上四分位数值要除以所有样品中上四分位数值的中位数。
以上三种方法效果大同小异,通常比较流行的是TMM和DESeq normalization
CPM 按照基因或转录本长度归一化后的表达即 RPKM (Reads Counts Per Million)、FPKM (Fragments Per Kilobase Million)和 TPM (Trans Per Million),推荐使用TPM######

1)直接选某个值过滤
keep <- rowSums(dgelist$counts) >= 50
dgelist <- dgelist[keep, ,keep.lib.sizes = FALSE]
2)利用cpm过滤
keep <- rowSums(cpm(dgelist) > 1 ) >= 2
dgelist <- dgelist[keep, ,keep.lib.sizes = FALSE]
实际的数据分析中,还需多加尝试,选择一个合适的过滤条件
3. 标准化
calcNormFactors()函数对数据标准化,以消除由于样品制备或建库测序过程中带来的影响。
这里选的是TMM标准化方法,还有其他的可以?calcNormFactors进行查看

TMM法
dgelist_norm <- calcNormFactors(dgelist, method = 'TMM')

dgelist_norm
plotMDS()是limma包中的方法,绘制MDS图,使用无监督聚类方法展示出了样品间的相似性(或差异)。可据此查看各样本是否能够很好地按照分组聚类,评估试验效果,判别离群点,追踪误差的来源等。
plotMDS(dgelist_norm, col = rep(c('red', 'blue'), each = 3))
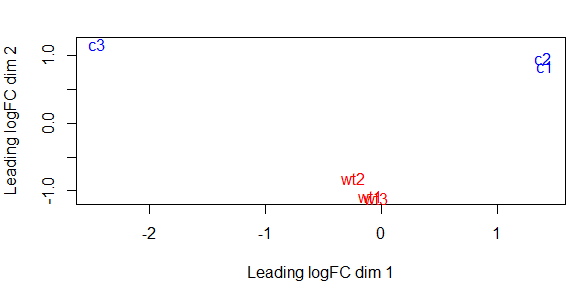
可以考虑一下出现较大偏差的原因
4. 估算离散值
负二项分布(negative binomial,NB)模型需要均值和离散值两个参数。edgeR中,分组矩阵使用model.matrix()获得,并可以通过estimateDisp()估算离散值。
design <- model.matrix(~group) #构建分组矩阵
dge <- estimateDisp(dgelist_norm, design, robust = TRUE) #估算离散值
plotBCV(dge) #作图查看

design中用0和1表示是哪一组,比如第二列1表示属于c2组
❗需要注意,标识好
0、1类型后,后面的差异分析将以分组1的基因表达量相较于分组0是上调还是下调为准进行统计。因此在本示例中,后续分析获得的基因表达量上调/下调均为分组c2相较于分组c1而言的。实际的分析中,切记不要搞反了。(有时会出现两组顺序相反的情况,还没找到方法怎么实现)
estimateDisp()实际上是个组合函数,可以一步得到多个计算结果,例如在上文我们使用分组矩阵design通过estimateDisp()估算了3个值,其实就是estimateGLMTagwiseDisp()、estimateGLMCommonDisp()和estimateGLMTrendedDisp()这3个结果的组合。如果不指定分组矩阵,则将得到estimateCommonDisp()和estimateTagwiseDisp()的结果组合。
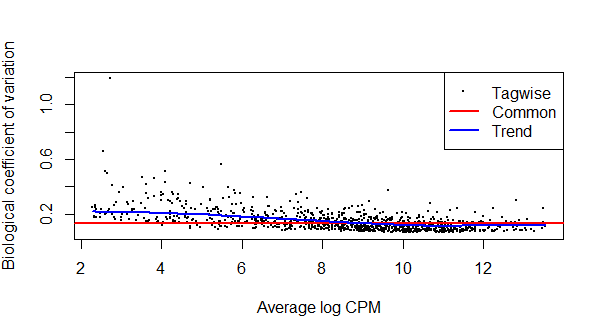
一定要记住是谁较谁
5. 差异基因分析
前面都是准备工作,现在可以开始正式分析了!
1) 负二项式广义对数线性模型(edgeR)
首先拟合负二项式广义对数线性模型(negative binomial generalized log-linear model),获取差异基因。这种方法大致可以这样理解,如果某个基因的表达值偏离这个分布模型,那么该基因即为差异表达基因。
使用edgeR包中的函数glmFit()和glmLRT()实现,其中glmFit()用于将每个基因的read count值拟合到模型中,glmLRT()用于对给定系数进行统计检验。
fit <- glmFit(dge, design, robust = TRUE) #拟合模 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章



















 609
609

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











