









DNA Sequencing Accuracy Comes a Long Way
PacBio strives to remove the choice between length and accuracy by optimizing their circular consensus sequencing method
By
-
August 12, 2019
Share
Since the beginning of the modern genomic sequencing era, researchers have had to make a choice. On the one hand there are highly accurate, short reads—the bread and butter sequences that come from Illumina and Complete Genomics. And on the other hand there are long reads, from Pacific Biosciences (PacBio) and Oxford Nanopore, that have notoriously suffered in accuracy.
The holy grail has been a platform that yields long, accurate reads. Today, the team at PacBio has published tweaks to their existing SMRT sequencing technology that brings them one step closer to it.
The work was recently published in Nature Biotechnology in a paper entitled, “Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome”.
“This is the first description of a method for generating read lengths that are both long and accurate,” notes Aaron Wenger, PhD, principal scientist in bioinformatics at PacBio and first author on the paper. The principle of PacBio’s circular consensus sequencing (CCS) has been around for a few years.
自从现代基因组测序时代开始,研究人员就不得不做出选择。
一方面,来自Illumina和Complete Genomics的面包和黄油序列具有高度精确的短序列。
另一方面,太平洋生物科学公司(PacBio)和牛津纳米孔公司(Oxford Nanopore)的长时间读取数据的准确性已经臭名昭著。
圣杯一直是一个平台,产生长,准确的解读。
今天,PacBio的团队发布了他们现有SMRT测序技术的调整,使他们离这个目标又近了一步。
这项工作最近发表在《自然-生物技术》上一篇题为《精确循环共识长读测序改进人类基因组变异检测和组装》的论文中。
PacBio生物信息学首席科学家、论文第一作者Aaron Wenger博士指出,这是第一种生成既长又准确读取长度的方法的描述。
PacBio的循环共识测序(CCS)的原理已经存在了好几年。
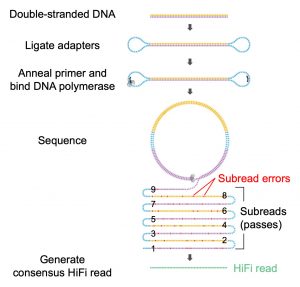
PacBio’s CCS system works by adding hairpin adapters ligated on each end of the linear DNA molecule, to create a “SMRTbell” template. The polymerase, bound to the adapter, moves from the adapter across the DNA insert, adding bases and creating the read of the sequence. CCS produces HiFi (high fidelity) reads by going around the adapter multiple times, continuing until the polymerase “dies” creating multiple passes of the same molecule. Typically, CCS is not considered a long-read technology. Wenger notes that the tradeoff to the platform’s high accuracy has been been reads that are 1000–2000-bases long. But, here, they have used the CCS approach to generate reads over 10,000 bases long.
How did they do it? Wenger says that one key was the technical innovation described in the paper known as “pre-extension”. Because PacBio sequencing relies on a camera taking frames, like in a movie, the polymerases are all independent of one another. They’ll keep adding nucleotides until they die and end the read.
The polymerases fall off for different reasons, commonly because the DNA is damaged. To this end, PacBio’s system is held captive to the quality of the DNA that is put on the instrument. So, they devised a way to minimize the amount of damaged DNA that gets placed on the instrument by selecting for undamaged DNA molecules. Wenger explains that they start the sequencing reaction before loading the DNA onto the instrument. After a few hours of extension, if the polymerase is still going, they conclude that the DNA is undamaged. Selectively loading that DNA onto the instrument is the key to getting long reads using their CCS method.
In addition to this, the researchers ensured that the selected DNA molecules are all about the same size by employing the SageELF instrument (made by Sage Science in Beverly, MA). Because they knew the size of the molecules, they knew the best duration for the pre-extension. This novel tweak was key to the process because it allowed the polymerase to go much farther once inside the sequencing instrument. This was “the last thing holding back the polymerase from stopping,” notes Wenger.
PacBio公司的CCS系统通过在线性DNA分子两端连接发夹适配器来创建SMRTbell模板。
结合到适配器上的聚合酶从适配器移动到DNA插入物上,添加碱基并创建序列的读取。
CCS通过在适配器周围多次循环来产生高保真度(HiFi)读数,直到聚合酶死亡,从而产生同一分子的多次传递。
通常,CCS不被认为是一种长读技术。
温格指出,该平台的高准确度的折衷已经读取了1000个2000个碱基的长度。
但是,在这里,他们已经使用CCS方法来产生超过10000个碱基长的数据。
他们是怎么做到的?
温格表示,其中一个关键是论文中所描述的技术创新,即所谓的“预扩展”。
因为PacBio测序依赖于摄像机拍摄帧,就像在电影中一样,聚合酶是相互独立的。
它们会不断添加核苷酸,直到它们死亡并结束阅读。
聚合酶由于不同的原因脱落,通常是因为DNA受损。
为此,PacBio系统受制于仪器上DNA的质量。
因此,他们设计了一种方法,通过选择未受损的DNA分子来减少放置在仪器上的受损DNA的数量。
温格解释说,他们在将DNA加载到仪器上之前就开始了测序反应。
延长几个小时后,如果聚合酶还在继续,他们就得出结论,DNA没有受损。
有选择地将DNA加载到仪器上是使用CCS方法获得长读数的关键。
除此之外,研究人员通过使用SageELF仪器(由马萨诸塞州贝弗利市的Sage Science公司制造)来确保所选的DNA分子大小相同。
因为他们知道分子的大小,所以他们知道预延伸的最佳持续时间。
这个新颖的调整是这个过程的关键,因为它允许聚合酶进入测序仪器后走得更远。
温格指出,这是阻止聚合酶停止的最后一件事。

Shawn Baker, PhD
PacBio reads typically have a really high error rate (~15% compared with ~0.1% for Illumina.) However, their errors tend to be random, so if the same region is sequenced several times, the errors average out resulting in a “consensus” sequence. Shawn Baker, PhD, consultant at SanDiegOmics.com explains it like this: “If you have a 1% error rate and you sequence to 100X depth, at each base you’ll get roughly 99 reads that look the same, say an ‘A’, and one base that is different, say a ‘G’. The consensus call would be to assume that the base is truly an ‘A’ and ignore the ‘G’ call.”
In this paper, PacBio showed that they can generate very high quality PacBio sequencing by reading the same molecule multiple times (~10 times on average) rather than comparing 10 separate reads together. This means that they end up with individual PacBio CCS reads with roughly the same error rate as Illumina reads, but which are much longer than Illumina reads.
In doing this, they generated highly accurate (99.8%), long, HiFi reads with an average length of 13.5 kilobases (kb). They applied the approach to sequence the well-characterized human HG002/NA24385 genome and obtained precision and recall rates of at least 99.91% for single-nucleotide variants (SNVs), 95.98% for insertions and deletions <50 bp (indels), and 95.99% for structural variants.
“The new CCS protocol together with the new 8M chips by PacBio are shown to be on par with short-read data from Illumina,” notes Albert Vilella, PhD, an independent consultant working with biotech companies in the U.K. Vilella tells GEN that “it is not yet a game-changer but an incremental improvement over the tools available to researchers and clinicians to assay genomes. It will allow assays to produce a more complete readout of all regions in the genome, including those with low mappability with short-reads technologies.”
Vilella continues that this “moves the field one step closer to being able to produce fully assembled complete individual genomes, for a similar cost as the current mapped-based short-read genomes.”
“It’s a big step forward” notes Keith Robison, PhD, principal scientist at Ginkgo Bioworks, because they are “interrogating more of the genome and giving haplotypes.”
Deanna Church, PhD, mammalian applications at Inscripta, raises the question of “polishing” and whether it would still be necessary with this PacBio approach. Polishing is the term given to the combined use of PacBio long reads with Illumina short reads. Typically, the short Illumina sequences are overlayed over long reads to polish them, or figure out where the errors are.
PacBio reads通常有非常高的错误率(~15%,而Illumina为~0.1%)。
然而,它们的误差往往是随机的,因此,如果对同一个区域进行多次排序,这些误差就会平均出来,从而得到一个一致的序列。
肖恩·贝克博士顾问在SanDiegOmics.com解释是这样的:如果你有1%的错误率序列100 x深度,你会得到大约99读取每个基地看起来一样的,说一个,,另一个基础是不同的,说一个G。
大家一致认为基数确实是A,忽略G值。
在这篇文章中,PacBio表明,他们可以通过多次读取同一个分子(平均大约10次)来产生非常高质量的PacBio测序,而不是将10个单独的reads放在一起比较。
这意味着他们最终得到的单个PacBio CCS的误差率与Illumina的读数大致相同,但比Illumina的读数要长得多。
在这样做的过程中,他们生成了高精确度(99.8%)、长、高分辨率的数据,平均长度为13.5 kb。
他们将这种方法应用于特征良好的人类HG002/NA24385基因组测序,获得单核苷酸变异(SNVs)的精确度和召回率至少为99.91%,插入和缺失50 bp (indels)的精确度和召回率为95.98%,结构变异的精确度和召回率为95.99%。
新CCS协议连同新8 m被PacBio显示芯片与短内容Illumina公司的数据,指出阿尔伯特•Vilella博士,一个独立的顾问与生物技术公司合作在英国Vilella告诉创尚不改变但增量改进工具分析基因组的研究人员和临床医生。
它将允许分析产生基因组中所有区域的更完整的读数,包括那些低定位能力的短读技术。
Vilella继续说,这使该领域向能够生产完全组装完整的单个基因组又迈进了一步,成本与目前基于mapped的短读基因组相似。
Ginkgo Bioworks首席科学家Keith Robison博士指出,这是一个巨大的进步,因为他们正在查询更多的基因组,并给出单倍型。
Inscripta的哺乳动物应用的Deanna Church博士提出了抛光的问题,以及这种PacBio方法是否仍然有必要进行抛光。
抛光是术语给予组合使用PacBio长读和Illumina短读。
通常情况下,短的Illumina序列被叠加在长序列上以修饰它们,或者找出错误所在。

Deanna M. Church, PhD
But Wenger asserts that this method eliminates the need for polishing. “The actual raw accuracy of these reads averages 99.8% which is similar to the accuracy of short reads.” Wenger points to a recent bioRxiv preprint from the Eichler lab, that looked at polishing HiFi reads with short reads and showed that polishing did not contribute increased accuracy.
Wenger notes that although the rate of mistakes is similar, the types of mistakes between short reads and HiFi reads are different. Baker adds that, as far as mistakes are concerned, indels are where Pacbio suffers—with higher indel error rates rather than base substitution errors. It’s “in the nature of how they do their sequencing,” he noted. The PacBio system captures those base additions as they happen; but they happen so quickly that the camera can miss it.
So, why wouldn’t everyone switch over to high-quality, long reads? Robison says that the main barrier is cost. “Each PacBio flowcell can deliver only so many reads, so you can choose to have lots of long, error-rich ones or fewer high-quality ones.” He notes that “if you are collecting 1/3 to 1/4 as many finished bases per flowcell but need 1/2 as many, you still need more flowcells to generate the data.” He adds that the question is to what degree labs will be willing to pay a premium and accept lower throughout for more variant information. Baker agrees that the cost per unit of data is going to go up. This method will “make PacBio more expensive per base generated, by reducing the data you get for the dollar.”
Who will use it, then? Wenger says that this is not going to be used for large-scale sequencing of populations, such as “sequencing everyone in Dubai.” Baker adds that people who choose this method will have to give up read length because “you won’t get that ultralong information that would help with, for example, phasing.”
So, it remains to be seen for which applications this method will be most useful. In general, Church thinks that the HiFi reads will be useful, but she also thinks that “you’ll still need some Oxford ultralong reads for good human assembly.” She also notes that “we need to get some good annotation data on these assemblies to know if you can do away with Illumina polishing.”
And, the competition is “not sitting idle,” notes Vilella. He adds that we could see similar multi-pass solutions from competitors soon. All of this will enable scientist to pick and choose the combination of technologies that gives them the best bang for their buck, such as combining long-reads and short-read polishing to produce high-quality end-to-end assembled genomes.
但是温格断言这种方法消除了修饰的需要。
这些读取的实际原始精度平均为99.8%,与短读取的精度相近。
温格指出了埃奇勒实验室最近的bioRxiv预印本,该预印本研究了用短读对HiFi进行抛光,结果表明抛光并没有提高精度。
温格指出,虽然错误率是相似的,但短读和高保真读的错误率是不同的。
Baker补充说,就错误而言,indels是Pacbio遭受更高indel错误率的地方,而不是碱基替换错误率。
他指出,这取决于它们如何进行测序。
PacBio系统捕捉这些碱基的添加;
但它们发生得如此之快,以至于摄像机无法捕捉到。
那么,为什么大家不转向高质量、长时间阅读呢?
罗宾逊表示,主要障碍是成本。
每个PacBio流式细胞仪只能传送这么多的数据,所以你可以选择有很多长的、错误丰富的数据,或者选择更少的高质量数据。
他指出,如果你在每个flowcell中收集1/3到1/4的已完成碱基,但需要1/2的碱基,你仍然需要更多的flowcell来生成数据。
他补充说,问题是,实验室愿意支付溢价到何种程度,愿意接受更低的价格来获取更多不同的信息。
贝克同意单位数据成本将会上升的观点。
这种方法将使PacBio每生成一个基更加昂贵,因为它减少了你花费一美元所获得的数据。
那么,谁会使用它呢?
温格说,这不会被用于大规模的人口排序,比如给迪拜的每个人排序。
贝克补充说,选择这种方法的人将不得不放弃阅读长度,因为你将无法获得帮助的超长信息,例如,相位。
因此,这种方法在哪些应用中最有用还有待观察。
总的来说,Church认为HiFi阅读将是有用的,但她也认为,你仍然需要一些牛津超长阅读,以良好的人类集会。
她还指出,我们需要获得这些组件的一些良好的注释数据,以知道是否可以取消Illumina抛光。
而且,Vilella指出,竞争并不是坐以待毙。
他补充说,我们很快就会看到竞争对手推出类似的多通道解决方案。
所有这些都将使科学家能够挑选和选择能给他们带来最大收益的技术组合,例如将长读和短读抛光结合起来,以产生高质量的端到端组装基因组。
















 2176
2176











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











