






 本文详细介绍了如何通过PubMed-Gene获取目标基因CDS序列,使用SnapGene软件分析酶切位点,并选择合适的双酶Kpn1和BamHI进行克隆。还涵盖了PCR扩增、酶切、连接、转化、质粒验证和测序等步骤,确保基因成功克隆到载体中。
本文详细介绍了如何通过PubMed-Gene获取目标基因CDS序列,使用SnapGene软件分析酶切位点,并选择合适的双酶Kpn1和BamHI进行克隆。还涵盖了PCR扩增、酶切、连接、转化、质粒验证和测序等步骤,确保基因成功克隆到载体中。
PubMed-Gene搜索需要克隆的基因


找到CDS区域,注意有的基因有很多isoform, 但大部分isoform的CDS区域相似,找常见或表达量较高的,或根据自己实验需要特定的isoform找到基因的CDS区域序列
打开SnapGene 软件-new DNA file, 粘贴CDS序列

分析其酶切位点


同时找到自己需要克隆载体的序列,分析多克隆位点(multiple cloning site, MCS)包含的酶切位点
找到MCS有的,而且不包括在克隆片段中的酶切位点
Kpn1 Acc65I BamHI NotI Xhol BstBI
按照在质粒的先后顺序,选择两个酶切位点https://www.neb.com/-/media/nebus/files/chart-image/cleavage_olignucleotides_old.pdf?la=en 或者https://wenku.baidu.com/view/0fb0ca1cb7360b4c2e3f644d.html?re=view 此网站有各个酶切位点应该选择的碱基情况
注意选择酶切效率高的位点
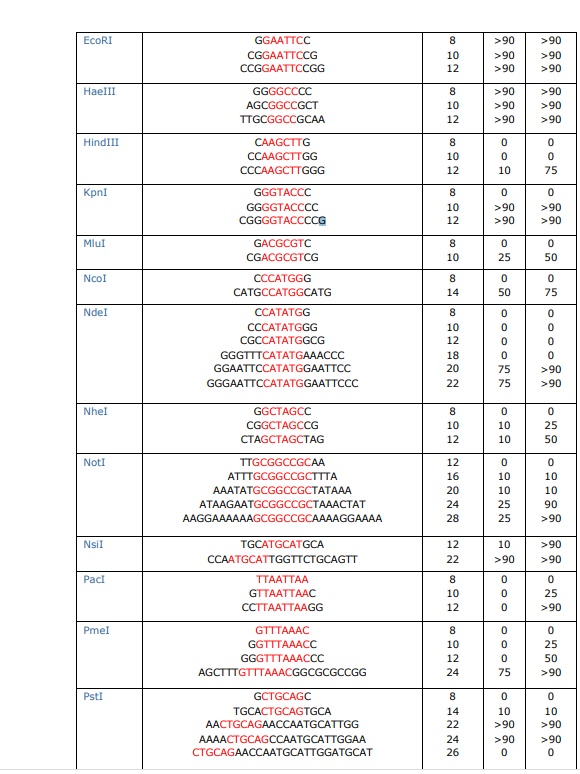
Kpn1 GGGGTACC
BamHI CGGGATCC
NotI ATAAGAATGCGGCCGC 这个必须20h以上酶切效率才能达到90%
Xhol CCGCTCGAG 20h只有75%
BstBI CTGCAGAACCAATGCATTGG 20h 90%
注意两个酶切位点不能选择切除后相同的回文序列,否则无法连接
选择合适的连个酶切位点后,评估所用buffer
https://nebcloner.neb.com/#!/redigest
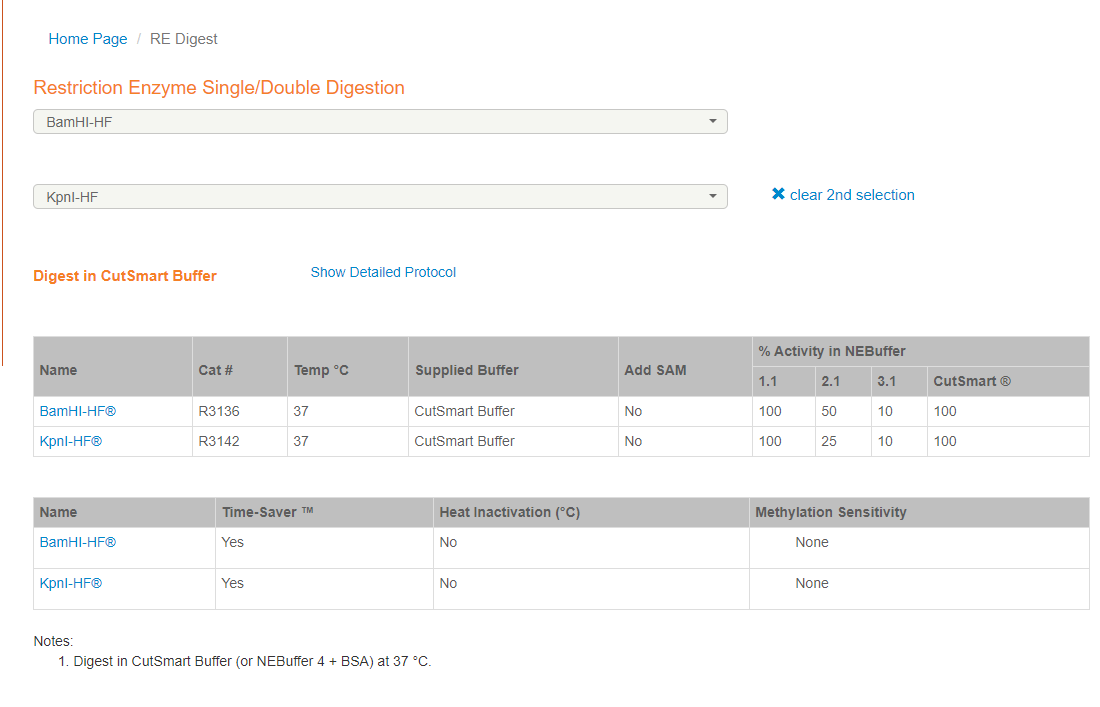
注意所选酶是HF酶还是普通酶,带星号表示不可用
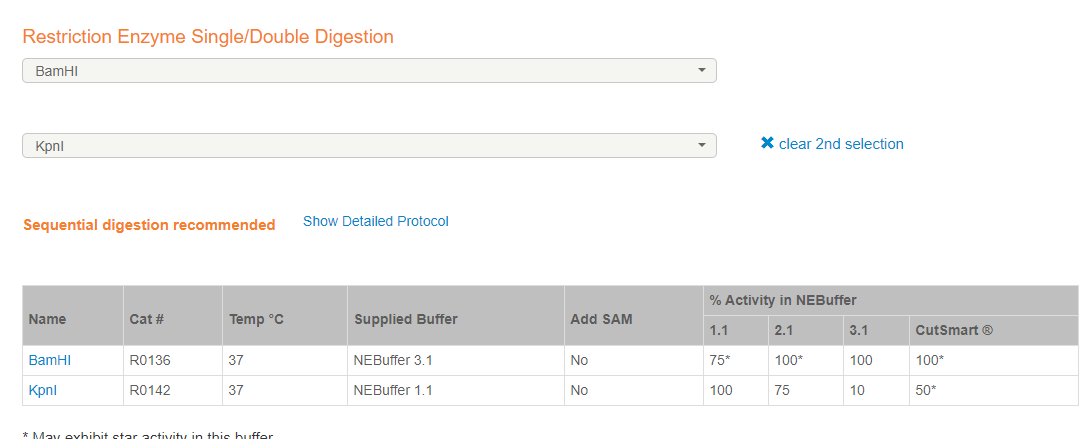
选择合适的双酶切系统后,设计引物,注意Vector的标记tag如果没有独立的启动子,则不应该将末尾3位终止密码子碱基克隆进去。
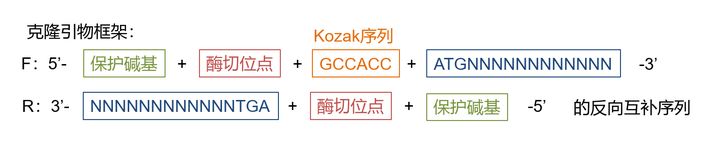
F:GGGGTACCGCCACCatgggccagactgggaagaaatc (KPNI)5-3
R: gcatcgaaagagaaatggaaatccctGGATCCGC (BamHI) 3-5
R: GCGGATCCagggatttccatttctctttcgatgc 5-3
2 PCR扩增目的基因
Mix(Thermo Phusion Master with HF)
7.5ul
2.5ul
DDW
6ul
19 ul
Primer(F+R, 浓度为5uM)
1
5
质粒>10ng,cDNA50-100ng
0.5ul
1
15ul体系
50ul体系

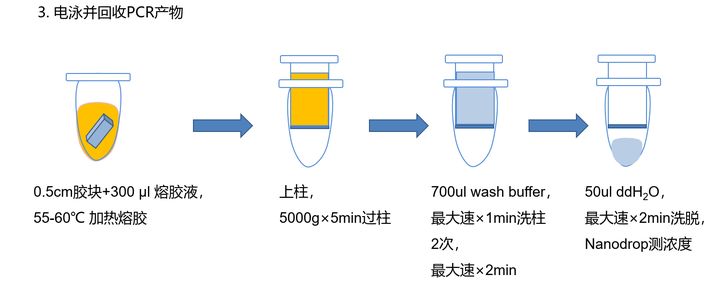
4. 酶切载体和PCR产物
PCR回收产物
34ul
Vector:1ug/浓度+DDW=34ul
Enzyme1
1ul
1ul
Enzyme2
1ul
1ul
10x smart cut buffer
4ul
4ul
40ul体系
40ul
37℃酶切过夜
5. PCR目的基因纯化
50ul体系加50ul DDW
100ul Binding buffer
Maximum 离心1min
700ulwash buffer
Maximum 离心1min
再Maximum 离心1min
35ul DDW洗脱
至于Vector则需要跑胶后根据质粒大小片段回收。
6.计算链接体系
PCR(ng)=7x30xPCR长度/载体大小长度
PCR:Vector 摩尔数之比大概1:7
连接体系
T4 ligase NEB M202s
1ul
10x T4 ligase buffer
2ul
DDW+PCR产物
7ul
Vector
Y ul (100ng/n)
20ul
酶切连接,室温>2h,最好16℃酶切过夜
T4说明书
COMPONENT
20 μl REACTION
T4 DNA Ligase Buffer (10X)*
2 μl
Vector DNA (4 kb)
50 ng (0.020 pmol)
Insert DNA (1 kb)
37.5 ng (0.060 pmol)
Nuclease-free water
to 20 μl
T4 DNA Ligase
1 μl
夏云方案:(xxx ug * 10e6) / (660pg * bp数)=xxx pmol
Vector 摩尔数 0.03 pmol(固定)0.03x0.6xVector(kb)/浓度(ng/ul)
DNA 摩尔数 乘以7
7. 转化

之后挑单克隆,摇菌(5ml 4管),12-16h后提取质粒
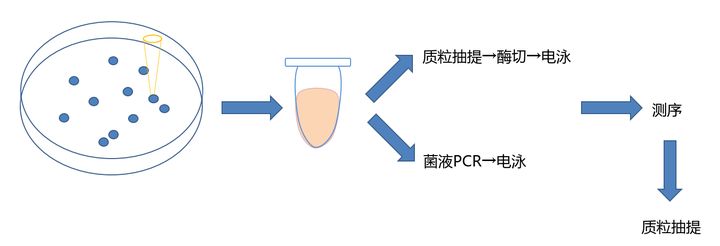
8. 跑胶验证质粒大小 140v 40min, 1-2%琼脂糖凝胶
1.正常质粒大小
2.酶切质粒
9. 测序,序列正确可以储存菌液 -80℃ 500ml菌液+500ml 40% glycerol 1:1稀释,终浓度为20%
PCR测序取的样本量
1-500bp
取1-10ng
500-1000bp
取5-20ng
1-2kb
取10-40ng
>2kb
取10-40ng
Plasmid测序取的样本量
1-5kb
取150-300 ng
Big size
取500-750 ng
DNA/plasmid
取约200ng体积
单引物(5uM)
1ul
DDW
总体系
6ul
注意测序大小只有600-800bp,如果测序片段较大,需要前后不同引物各测序一次
10 转染
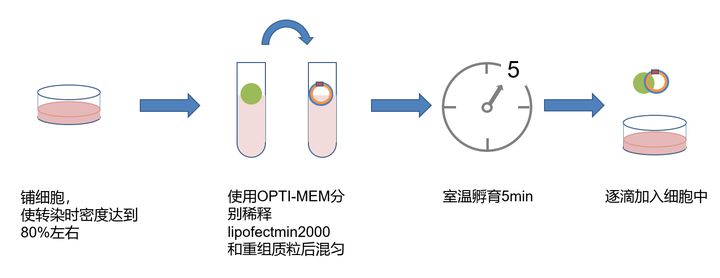
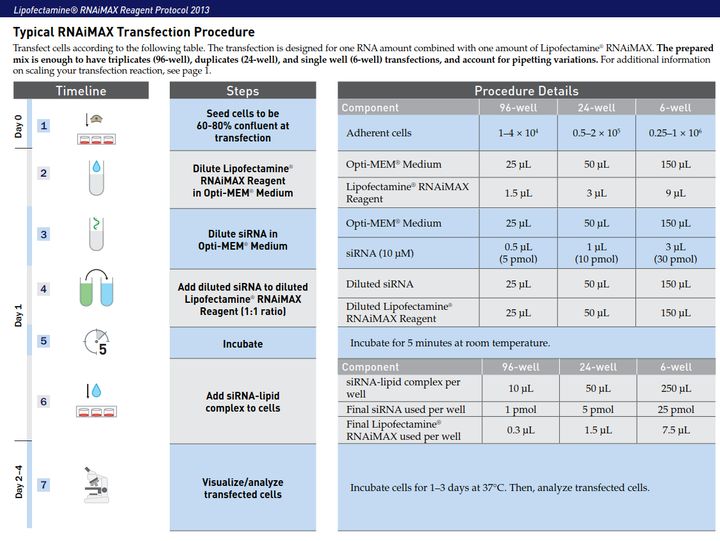
11. qPCR及WB验证目的基因是否过表达

















 2651
2651

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









