






 本文详述了单细胞转录组分析的步骤,包括预处理(质控、归一化、数据矫正)、细胞层面的下游分析(细胞分群、轨迹分析)和基因层面的分析。强调了数据矫正与整合的重要性,以及在细胞分群和轨迹推断中的策略。同时,讨论了差异表达分析、基因集分析和基因调控网络在理解细胞异质性中的作用。文章提供了最佳实践和工具推荐,适合新老研究者参考。
本文详述了单细胞转录组分析的步骤,包括预处理(质控、归一化、数据矫正)、细胞层面的下游分析(细胞分群、轨迹分析)和基因层面的分析。强调了数据矫正与整合的重要性,以及在细胞分群和轨迹推断中的策略。同时,讨论了差异表达分析、基因集分析和基因调控网络在理解细胞异质性中的作用。文章提供了最佳实践和工具推荐,适合新老研究者参考。

 今天是生信星球陪你的第618天
今天是生信星球陪你的第618天
大神一句话,菜鸟跑半年。我不是大神,但我可以缩短你走弯路的半年~
就像歌儿唱的那样,如果你不知道该往哪儿走,就留在这学点生信好不好~
这里有豆豆和花花的学习历程,从新手到进阶,生信路上有你有我!
刘小泽写于2020.5.6-5.8
历时三天,终于理解完!不仅仅是综述,更不是纯翻译文,而是将其中重要的知识点和之前个人所学结合起来”一阳指和狮吼功合并为一整招“
最后,强烈建议打开原文链接,因为所有文献都是有链接的,可以直接点进去
1 文章信息
题目:Current best practices in single-cell RNA-seq analysis: a tutorial
发表日期:2019年6月19日
杂志:Mol Syst Biol
文章在:https://www.embopress.org/doi/10.15252/msb.20188746
DOI:https://doi.org/10.15252/msb.20188746

2 摘要
单细胞领域日新月异,大量的工具被开发出来,但很难去判断是否好用,而且如何组建一个分析流程是一个难点。本文将详细介绍单细胞转录组数据分析的步骤,包括预处理(质控、归一化标准化、数据矫正、挑选基因、降维)以及细胞和基因层面的下游分析。并且作者将整个流程应用在了一个公共数据集作为展示(详细说明在:https://www.github.com/theislab/single-cell-tutorial),目的是帮助新入坑用户建立一个知识体系,已入坑用户更新知识体系。
3 前言
需要注意,虽然在原文链接中这些文献链接可以打开,但会链接到原文的该文献位置,而不是直接打开该文献
现在已经可以利用scRNA研究斑马鱼、青蛙、涡虫的细胞异质性(Briggs et al, 2018; Plass et al, 2018; Wagner et al, 2018) ,重新理解以前的细胞群体,但这个领域面临的一个问题就是没有成熟的标准化流程。标准化之路的困难有:大量分析方法和工具的诞生(截止2019.3.7 已经有385种工具)、爆炸式增长的数据量(Angerer et al, 2017; Zappia et al, 2018)。另外根据不同研究目的,各种分支也突显,例如在细胞分化过程中预测细胞命运(La Manno et al, 2018)。在我们眼界大开的同时,分析流程标准化就变得更加困难。
在未来分析流程标准化之路上,困难还会存在于技术整合层面。比如现在大量的scRNA工具都是用R和Python写的,跨平台分析需求在增长,而对编程语言的喜好也决定了工具的选择。很多好用的分析工具将自己限制在用各自的编程语言开发的环境中,例如Seurat、Scater、Scanpy。
接下来,就一起看看作者列出了哪些他认为比较好的软件和流程吧
先上一个scRNA分析总体流程图:
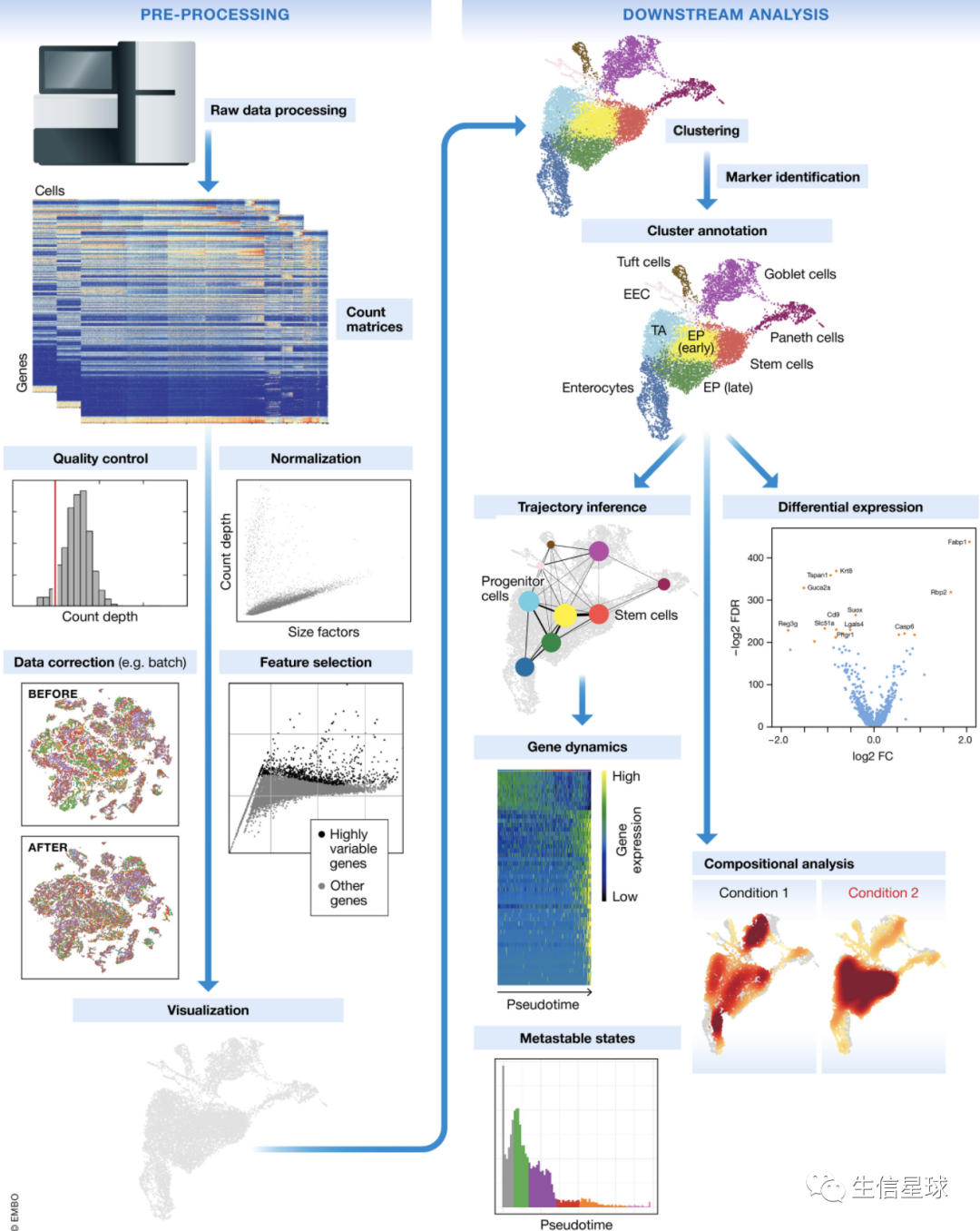
4 预处理和可视化
4.1 首先看一下实验过程
比较详细的介绍可以看:Ziegenhain et al (2017); Macosko et al (2015); Svensson et al (2017).
原文描述的关键点是:
4步走:Typical workflows incorporate single‐cell dissociation, single‐cell isolation, library construction, and sequencing.
组织裂解=》细胞分离=》文库构建=》测序第一步:As a first step, a single‐cell suspension is generated in a process called single‐cell dissociation in which the tissue is digested.
第二步:To profile the mRNA in each cell separately, cells must be isolated. 写了主要的2种方法:plate‐based、droplet‐based,当然也都存在一些问题:In both cases, errors can occur that lead to multiple cells being captured together (doublets or multiplets), non‐viable cells being captured, or no cell being captured at all (empty droplets/wells)
第三步:Each well or droplet contains the necessary chemicals to break down the cell membranes and perform library construction. Furthermore, many experimental protocols also label captured molecules with a unique molecular identifier (UMI).
UMI的作用主要是区分:UMIs allow us to distinguish between amplified copies of the same mRNA molecule and reads from separate mRNA molecules transcribed from the same gene.
第四步:Libraries are labelled with cellular barcodes and pooled together (multiplexed) for sequencing.
感觉原文描述的还没有illumina给出的详细,那么就看看illumina的图文并茂版:
illumina单细胞测序工作流程:关键步骤和注意事项:http://web.illumina.com.cn/landing/products_view.asp?newsid=324
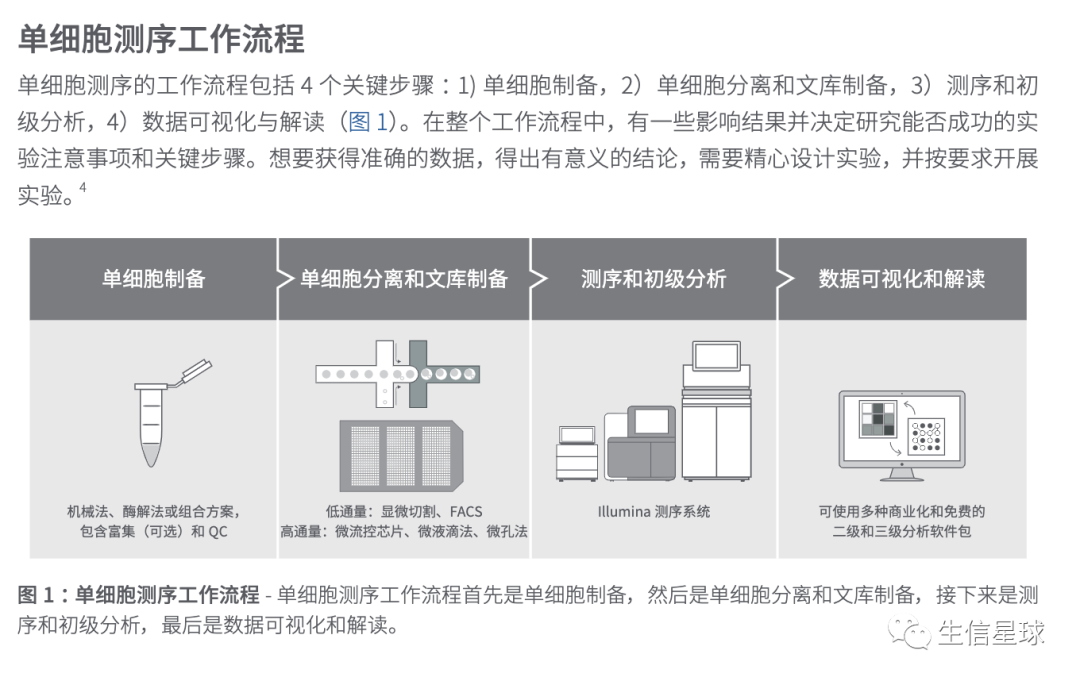
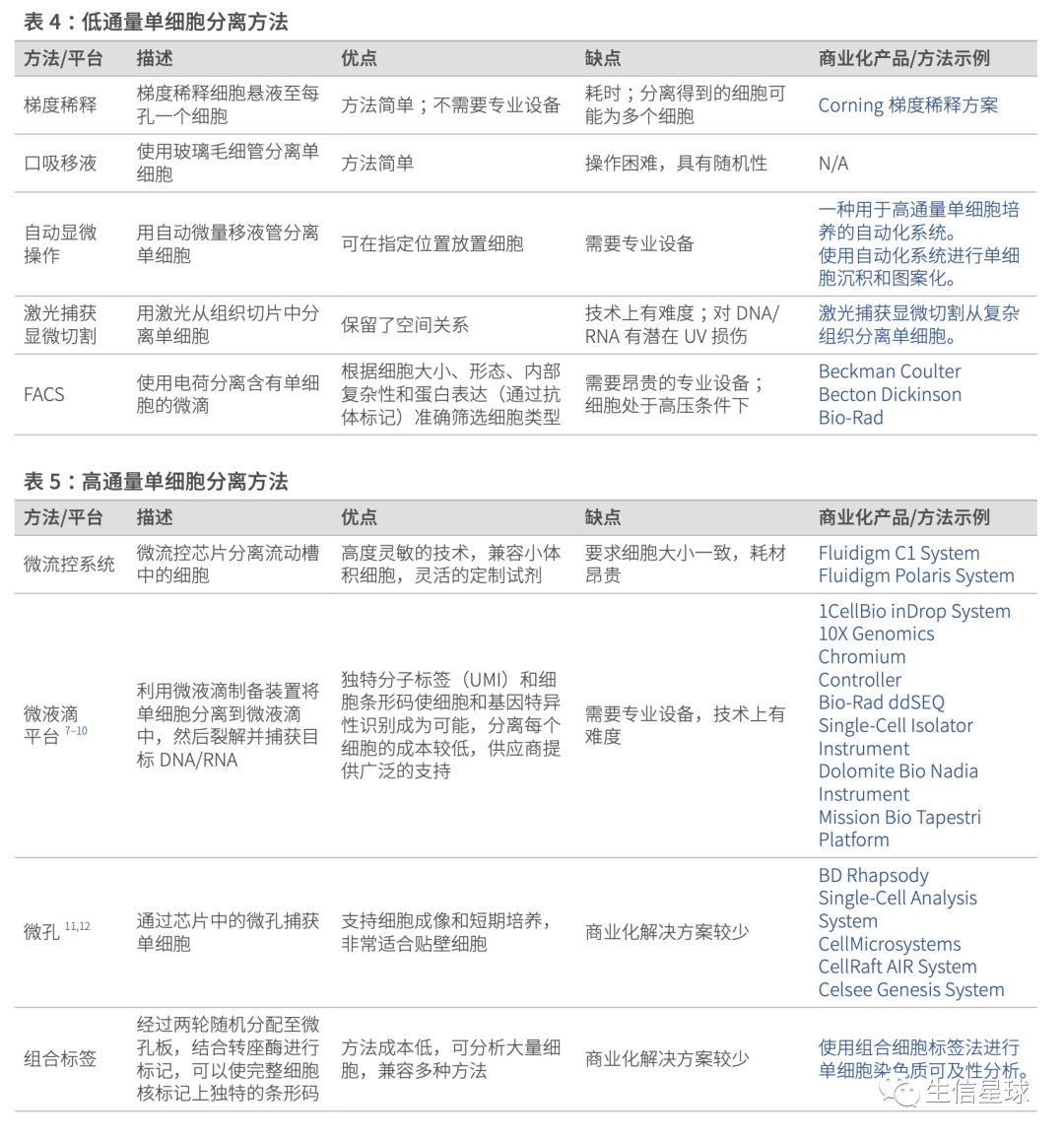
原始测序数据要经过处理得到表达矩阵,注意这里有两种表述方式:molecular counts (count matrices) 【也即是使用UMI的】和 read counts (read matrices),取决于是否使用UMI。而作者介绍的流程中,默认使用 count matrices,除非readmatrices和 count matrices得到的结果存在差异,才会特别介绍read matrices
关于比较read and molecule counts,有人写了一个R流程:https://jdblischak.github.io/singleCellSeq/analysis/compare-reads-v-molecules.html
原始数据处理工具主要有:CellRanger、indrops、SEQC、zUMIs
它们主要做了这么几件事:
read quality control (QC)
assigning reads to their cellular barcodes and mRNA molecules of origin (also called “demultiplexing”)
genome alignment
quantification
得到的矩阵行是转录本,列是barcodes【这里用barcodes而不是直接叫细胞,是因为不同细胞的reads也可能属于同一个barcode =》如果出现一孔/液滴多细胞(doublet情况),那么barcode在多个细胞都是一样的】当然也会出现有barcode但实际没有细胞的情况(一个孔/液滴没有细胞即droplet,但这个孔/液滴也会赋予barcode)
反正记住:barcode和孔/液滴是对应的,但一个孔/液滴中有一个细胞还是多个细胞或者没有细胞,都会存在barcode。只不过最后可能会看到:多个细胞对应一个barcode、即使没有细胞也会有barcode这样的情况
关于10X实验环节,可以看我之前写的:知根知底才能有的放矢

 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章
















 447
447











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









