






计算大牛David Baker等人于2022年8月30日在Cell上发表了一篇“Accurate de novo design of membrane-traversing macrocycles”。下面我们来做一下翻译和解读。
简要介绍
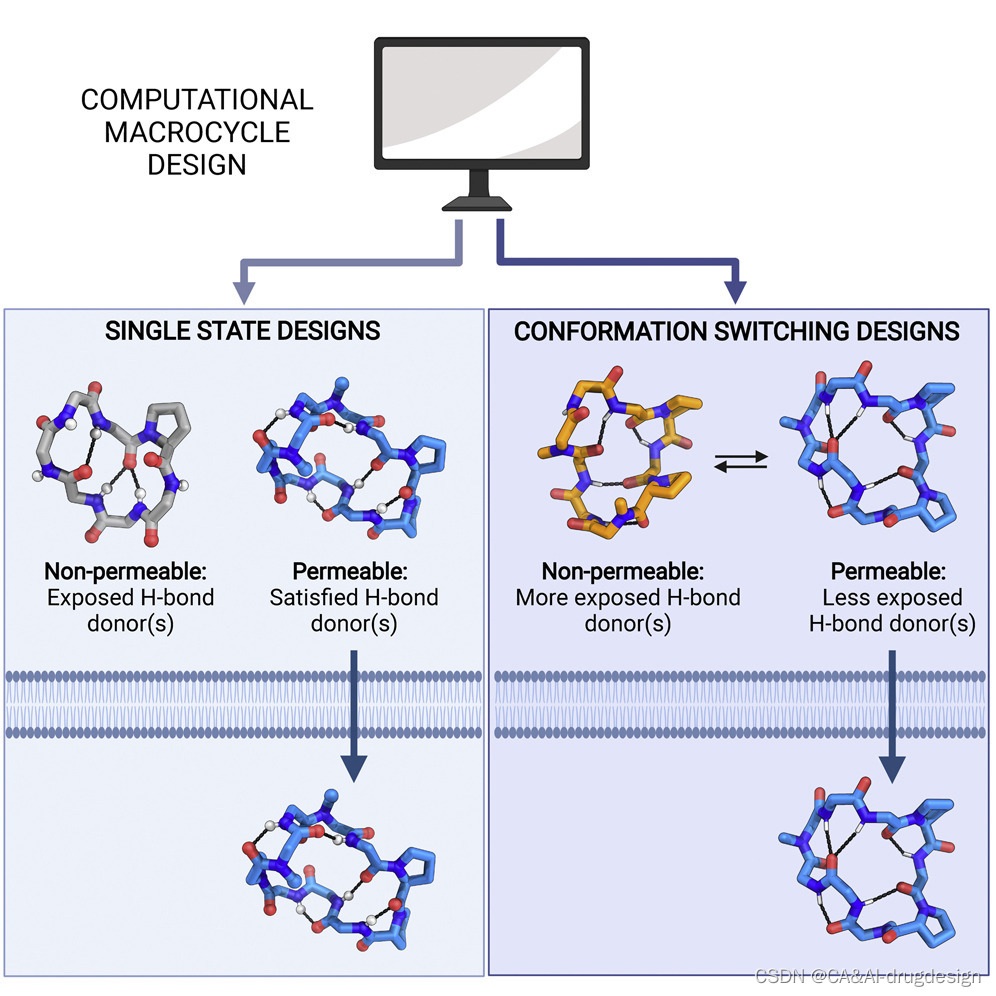
对大环肽穿膜和口服生物利用度的设计原则的探索,使产生的合成大环肽能够折叠成预测的构象,能够穿过膜,甚至能够根据极性与非极性的环境采取不同的构象。
亮点
- 计算设计超越 "五项规则 "空间的多样化的穿膜大环肽;
- 设计的大环肽的X射线和核磁共振结构与它们的计算模型相匹配;
- 设计的大环肽在体外具有穿膜性,在体内具有口服生物利用;
- 设计的变色龙肽(chameleonic被定义为长度为5或6个残基的α螺旋或β折叠肽)显示了溶剂依赖性的构象转换;
图片摘要
摘要
我们利用计算设计和实验表征,系统地研究了大环肽穿膜和口服生物利用度的设计原则。我们设计了184个含有6-12个残基的大环肽,其预测结构范围很广,含有非经典的骨架修饰,实验确定的结构有35个;这35个中,29个大环肽的结构与计算模型非常接近。通过这样的控制,我们表明,通过确保所有的酰胺(NH)基团参与内部氢键相互作用,可以系统地获得大环肽的穿膜属性。在6-12个残基范围内的84种设计的大环肽可以穿过膜,其表观渗透率大于1×106cm/s。通过设计脂质膜中喜欢的另一种等能的完全氢键状态,可以使具有暴露的NH基团的大环肽具有穿膜性。能够稳定地设计具有高结构准确度、能穿膜和口服生物利用度高的多肽,应该有助于设计下一代的大环肽疗法。
引言
大环肽作为治疗药物具有相当大的潜力,在破坏蛋白-蛋白相互作用的能力方面比小分子有优势,在代谢稳定性和跨膜能力方面比蛋白质有优势(Muttenthaler et al., 2021)。具有穿膜属性的肽可以进入细胞内的药物靶点,通过肠道上皮细胞转运,实现口服给药,并通过穿越脑微血管内皮细胞穿透血脑屏障。自然出现的大环肽,如环孢菌素A和格列宁霉素,表明大环肽穿膜的潜在机制超出了传统的 “五规则”(Ro5)(Bockus and Lokey, 2017; Bockus et al., 2015; Hewitt et al., 2015; Nielsen et al., 2017)。然而,事实证明,对少数自然穿膜大环肽的了解,并将其用于开发不同形状和大小的新的穿膜肽是非常困难的。对天然可穿膜的大环肽的研究发现了一些共同的特征,如低极性表面积和缺乏不如意的氢键供体(Bockus et al., 2015; Peraro and Kritzer, 2018; Rand et al., 2012; Rezai et al., 2006)。在水和膜环境中不同结构之间的构象转换也被描述为实现被动穿膜的一种潜在方法(Peraro and Kritzer, 2018; Wang et al., 2018; White et al., 2011)。将这些特征纳入基于文库的方法,使得一些新的穿膜环肽的开发成为可能。然而,以前的工作主要局限于较小的肽的情况,通常是5-7个氨基酸(Hewitt et al., 2015; Hill et al., 2014; Wang et al., 2014; White et al., 2011)。此外,将这些原则扩展到具有广泛化学和结构多样性的新型穿膜肽的设计上仍然具有挑战性,因为它需要了解结构、灵活性和穿膜性之间的关系,以及同时准确控制大环肽的序列和结构特征的能力(Mulligan,2020)。
在此,我们利用计算设计的能力来指定大环肽的结构,系统地探索被动穿膜的决定因素(Bhardwaj et al., 2016; Hosseinzadeh et al., 2017)。我们采用设计-构建-测试的方法,设计含有不同结构特征的肽,确定其晶体和溶液结构,并评估其穿膜性。**我们考虑三个结构特征:**通过形成肽内氢键满足所有氢键供体,存在顺式肽键,以及在水和脂质环境中切换构象的能力。
结果
我们首先研究了所设计的具有广泛不同长度和结构的、并且仅具有“所有NH基团完全内部满足”特性的大环肽是否能稳定地穿越脂质膜。我们扩展了Rosetta generalized kinematic closure(genKIC)方法,随机生成了~106个6-12个残基的聚甘氨酸肽的N-C环状骨架构象的集合,通过从平底对称的Ramachandran表中随机选择phi/psi torsions来采样环状骨架构象(Bhardwaj et al., 2016; Hosseinzadeh et al., 2017)。从这些大的数据集中,我们选择了至少有两个分子内氢键的骨架,并进行了Rosetta组合序列设计,将L-和D-氨基酸分别限制在Ramachandran空间的负phi区和正phi区,并在结构上兼容的位置加入构象受限的氨基酸,如L-脯氨酸、D-脯氨酸和α-氨基异丁酸(AIB)。为了清除暴露的和不令人满意的氢键供体,骨架上有不令人满意的氢键供体的氨基酸被突变为其N-甲基化的变体,并且在序列设计步骤中只允许使用非极性氨基酸(见STAR方法和图S1A)。我们选择了具有两个或更多分子内氢键和五个或更少N-甲基化氨基酸的低能量设计。通过生成105-106个备选构象并评估能量和骨架RMSD与设计模型的关系,对所选大环肽的构象能量景观(landscape)进行了表征。我们选择了具有漏斗状能量景观(landscape)的序列,并向其相应的设计模型靠拢(见STAR方法和图S1B)。使用基于骨架扭转角的聚类方法(Hosseinza- deh et al., 2017)评估了整体结构的多样性。每个残基都被分配了一个torsion bin(A[右手螺旋区],B[右手折叠区],X[A的镜像],Y[B的镜像],O[残基i和i+1之间具有phi<0和顺式肽键的氨基酸]和Z[残基i和i+1之间具有phi>0和顺式肽键的氨基酸]),并对由此产生的torsion bin strings(例如,对于一个六残基的肽,XYABOX)进行聚类。因为在一个环中,启点残基的选择是随机的,而且被动的穿膜性和折叠倾向对镜像是不变的,所以将具有在循环排列或镜像反转下相互转化的bin strings的聚类合并,并选择所产生的非生成clusters的成员(我们用在所有排列和反转中扭转bin string的最低字母顺序表示)进行化学合成和实验特征分析。
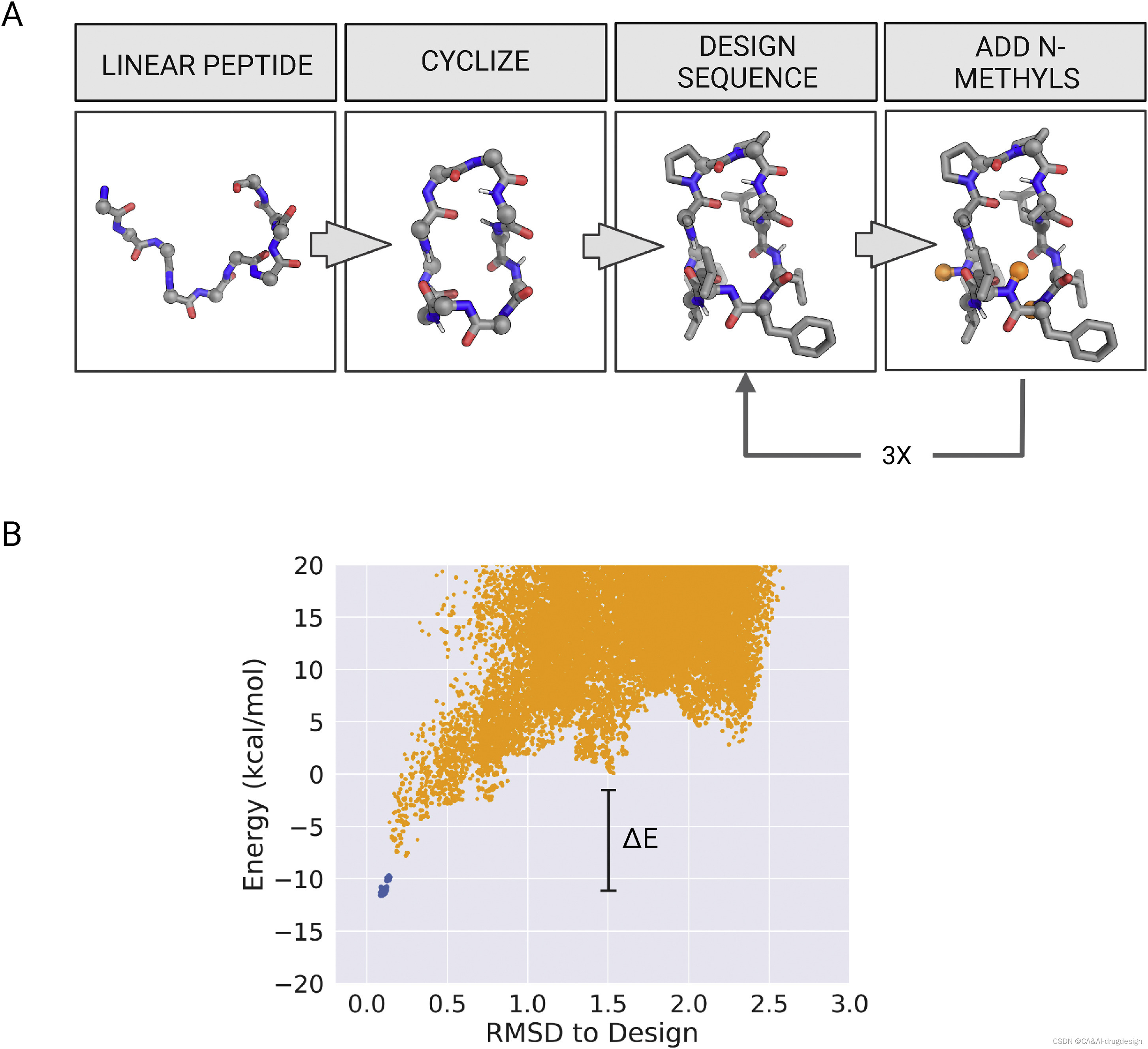
图S1 穿膜多肽的设计和选择,与图1和图3以及STAR方法有关。
6-8个残基设计的大环化合物的穿膜性
我们首先测试了我们的设计管道在6-8个残基大环肽上控制大环结构和穿膜性的能力,这些大环肽包括不同的结构和N-甲基化模式。对于6、7和8个残基的大环肽,我们分别选择了8、5和19种设计(代表6、5和16个聚类),这些设计具有完全满意的骨架NH基团和漏斗状的能量景观,并且跨越了不同的序列、结构、N-甲基化模式和结构片段(序列和设计模型见Data S1和S2)。选定的大环肽是用化学方法合成的,并用反相高效液相色谱法(RP-HPLC)进行纯化。在某些情况下,观察到两个具有预期分子量的峰;在这种情况下,我们通过实验分别测试这两个峰,并在设计名称中把它们称为p1和p2。
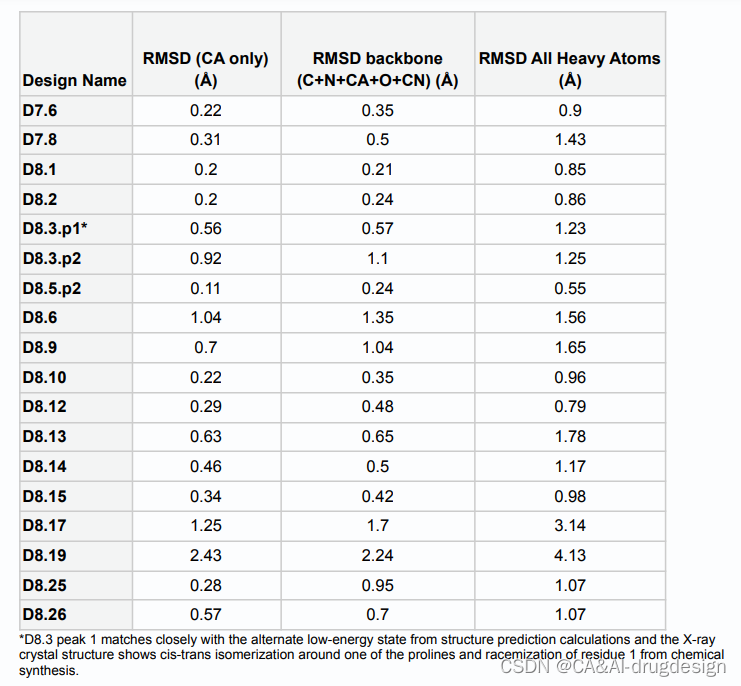
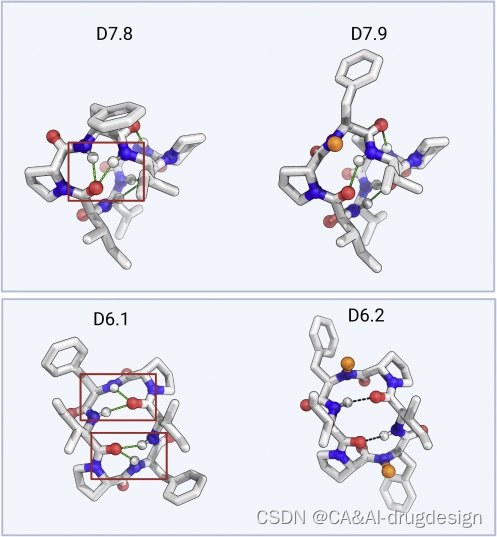
为了评估设计方法的准确性,我们确定了两个7残基大环肽和15个8残基大环肽的X射线晶体结构。两个7残基和12个8残基大环肽的结构与计算设计模型非常接近(骨架原子RMSD<1.2 A˚)(图1;Table S1)。在七个RMSD明显低于0.5 A˚的情况下,设计模型在X射线数据的实验分辨率之内。其中三个设计,D8.1,D8.2,和D8.12,是内部对称的:D8.1的骨架与设计模型的RMSD为0.21 A˚,其内部S2镜像对称性由4个β turns和2个γ turns motif组成;D8.2的C2对称性由4个β turns和2个α turns组成(RMSD为0.24 A˚);D8.12的C2对称性有4个内部氢键,没有N-甲基化氨基酸(RMSD为0.48 A˚)(图1)。D8.5.p2的设计没有N-甲基化的氨基酸,通过四个内部氢键形成四个β turns和一个α turn的motif来稳定(RMSD 0.24 A˚ )。D8.10的模型和X射线结构之间的RMSD为0.35 A˚,它的序列中有三个N-甲基化氨基酸和两个脯氨酸残基;它的其他三个氨基酸参与了三个内部氢键。D7.6的特点是有三个β turns和一个N-甲基化氨基酸,与设计出来的结构非常接近(RMSD 0.35 A˚)。D7.8有五个内部氢键,没有N-甲基化氨基酸(RMSD 0.5 A˚)。总的来说,实验结构和设计模型之间的密切匹配验证了我们的方法可以非常准确地指定大环肽结构(表S1)。
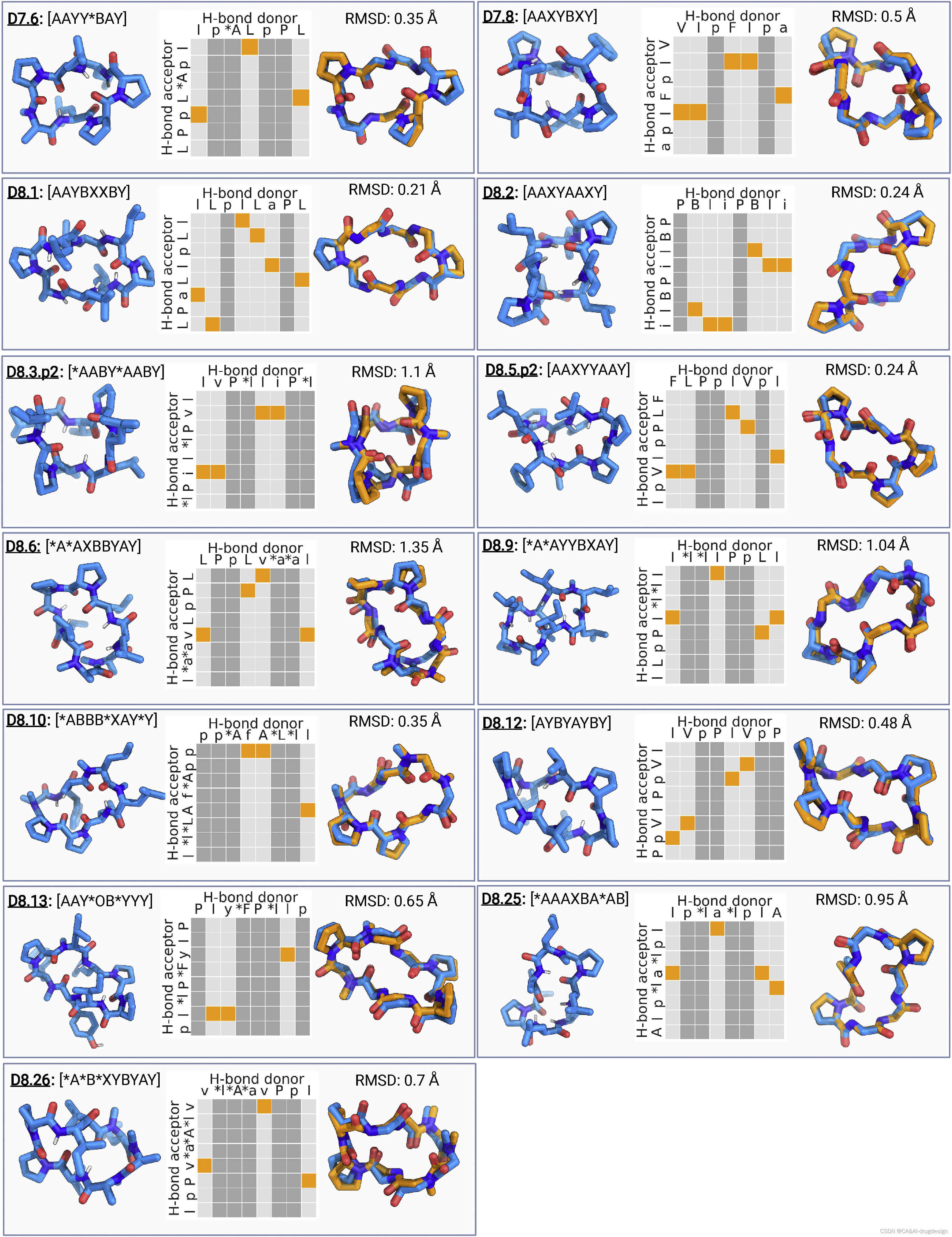
图1 6-8个氨基酸大环化合物的计算设计和结构验证
表S1. 与图1相关的7-和8-氨基酸大环肽的设计模型和X射线晶体结构的RMSD
在发现大环肽按设计折叠后,我们接下来用transwell permeability assays探索了它们的穿膜性。在平行人工膜渗透试验(PAMPAs)(Di et al., 2003)中,穿越人工膜的速率是通过基于质谱技术的供体和受体孔中肽浓度的量化来确定的(见STAR方法)。8个6-聚体、5个7-聚体和16个8-聚体的表观渗透率(Papp)大于1×10-7(见Data S3)。其中,8个6聚体、5个7聚体和10个8聚体的Papp大于1×10-6cm/s(图2A)。PAMPA是衡量被动跨脂质膜转运的一个很好的指标;然而,体内的口服生物利用度涉及到外排转运体的细胞屏障。因此,我们在transwell Caco-2 assays中测试了所设计的大环肽的一个子集,以结直肠上皮细胞作为供体和受体孔之间的屏障,并在外排转运的相反方向。在Caco-2试验中,渗透性大于1×10-6cm/s被认为是候选药物的充分穿膜的标志。由于Caco-2试验需要大量资源,我们集中测试较大的8个残基的大环肽,而没有对6和7个残基的大环肽进行这种试验。我们再次观察到Caco-2试验中的高穿膜性(图2B):在Caco-2试验中测试的8个含有8个残基的大环肽中,6个大环肽的Papp大于1×10-6cm/s,4个大环肽高于1×10-5cm/s。没有N-甲基化氨基酸的D8.1显示出非常高的Papp,为23.27×10-6 cm/s。

图2 计算设计的大环肽在PAMPA和Caco-2试验中的穿膜性测量。
Papp大于1×10-6 cm/s和漏斗状的能量景观的穿膜大环肽涵盖了广泛的结构,6、7和8个残基的大环肽分别有6、5和9个聚类。虽然以前采用天然骨架和基于库的方法的研究已经鉴别了穿膜大环肽,但这些大环肽通常需要多个N-甲基化或N-烷基化的氨基酸。例如,环孢素中11个氨基酸中有7个是N-甲基化的,这是以增加分子灵活性和合成难度为代价的。在我们的6-8个残基穿膜肽中,N-甲基化氨基酸的数量在0到3之间。在结构验证的设计大环肽中,有5个大环肽,即D7.8、D8.1、D8.2、D8.5和D8.12,没有N-甲基氨基酸,突出了计算方法所提供的精确控制,通过内部氢键和脯氨酸设计出完全满足氢键供体的结构(图1;数据S2)。D8.1、D8.2、D8.5.p2和D8.12是我们所知的缺乏N-甲基化或N-烷基化氨基酸的最大被动穿膜大环肽。在这些没有N-甲基化氨基酸的大环肽中,广泛的内部氢键使NH键得到充分满足;D8.1和D8.2有六个内部氢键,使结构稳定。一些6个和7个残基的大环肽设计模型(D6.3、D6.5、D6.9和D7.8)包含一个重叠的β和γ turns的几何紧张排列(图S2),其中中间的氨基酸可能有一个部分或完全不满足的NH基。因此,我们也设计并测试了中间氨基酸被N-甲基化的变体,在所有情况下,具有额外N-甲基化的变体比原来的变体穿膜性更强。然而,在所有的穿膜大环肽中,穿膜性与N-甲基化氨基酸的数量无关;例如,D8.1是PAMPA和Caco-2试验中穿膜性最强的含有8个残基的大环肽,没有任何N-甲基化氨基酸。这说明在折叠结构中最大限度地满足氢键和少用N-甲基化是实现穿膜性的一个可行策略。
图S2 几何学上的strained turn类型的N-甲基化,与图1有关
穿膜的、9-12残基的大环肽的设计
早期关于被动穿膜肽的工作主要局限于5-7个氨基酸,有几个天然产物的较大的肽衍生物的案例(Hewitt et al., 2015; Hill et al., 2014; Wang et al., 2014; White et al., 2011)。这是因为纯亲脂多肽的穿膜性随大小而急剧下降,正如在对所有残基都被N-甲基化的8-mer、9-mer和10-mer多肽的研究中所观察到的(Pye et al., 2017)。为了确定我们的设计原则是否能规避这一趋势,我们使用我们的计算管道设计了各种较大的大环肽,范围从9到12个氨基酸。
我们选择合成和表征了17个9-mer、41个10-mer、19个11-mer和8个12-mer的大环肽,其漏斗状的能量景观分别跨越16、37、18和8个不同的结构聚类,结构范围广泛,N-甲基化氨基酸在1到6之间(序列和设计模型见Data files S1和S2)。50个设计的大环肽分别跨越12、23、13和2个结构聚类的9、10、11和12个残基,在PAMPA试验中具有穿膜性,表观渗透性大于1×10-7 cm/s(见数据S3)。其中,10个9-mers、16个10-mers、9个11-mers和1个12-mers在PAMPA试验中显示出明显的穿膜性(Papp>1×10-6 cm/s)(图2A)。它们的穿膜性有大小之分,但其下降幅度没有以前对非设计的大环肽的研究中观察到的那么陡峭,导致显著的穿膜性超出了可穿膜的类药化合物的典型大小范围(“五规则”,见介绍)。在Caco-2试验中,3个9-mer、8个10-mer、7个11-mer的大环肽和一个12-mer的大环肽的Papp大于1×10-6 cm/s(图2B)。尽管它们的尺寸很大,但多种10个和11个残基的大环肽在Caco-2试验中显示出相当高的Papp:D10.19、D10.30和D10.34的速率大于1×10-5 cm/s,另外三个设计的大环肽的Papp在0.5和1.0×10-5 cm/s之间。一个11-mer的大环肽,D11.3,在Caco-2试验中具有9.11×10-6 cm/s的高Papp(图2B;数据S3)。测试的12个残基的大环肽中的大多数不具有穿膜性,但D12.6在PAMPA和Caco-2试验中的Papp值分别为1.47×10-6 cm/s和2.84×10-6 cm/s(图2B)。
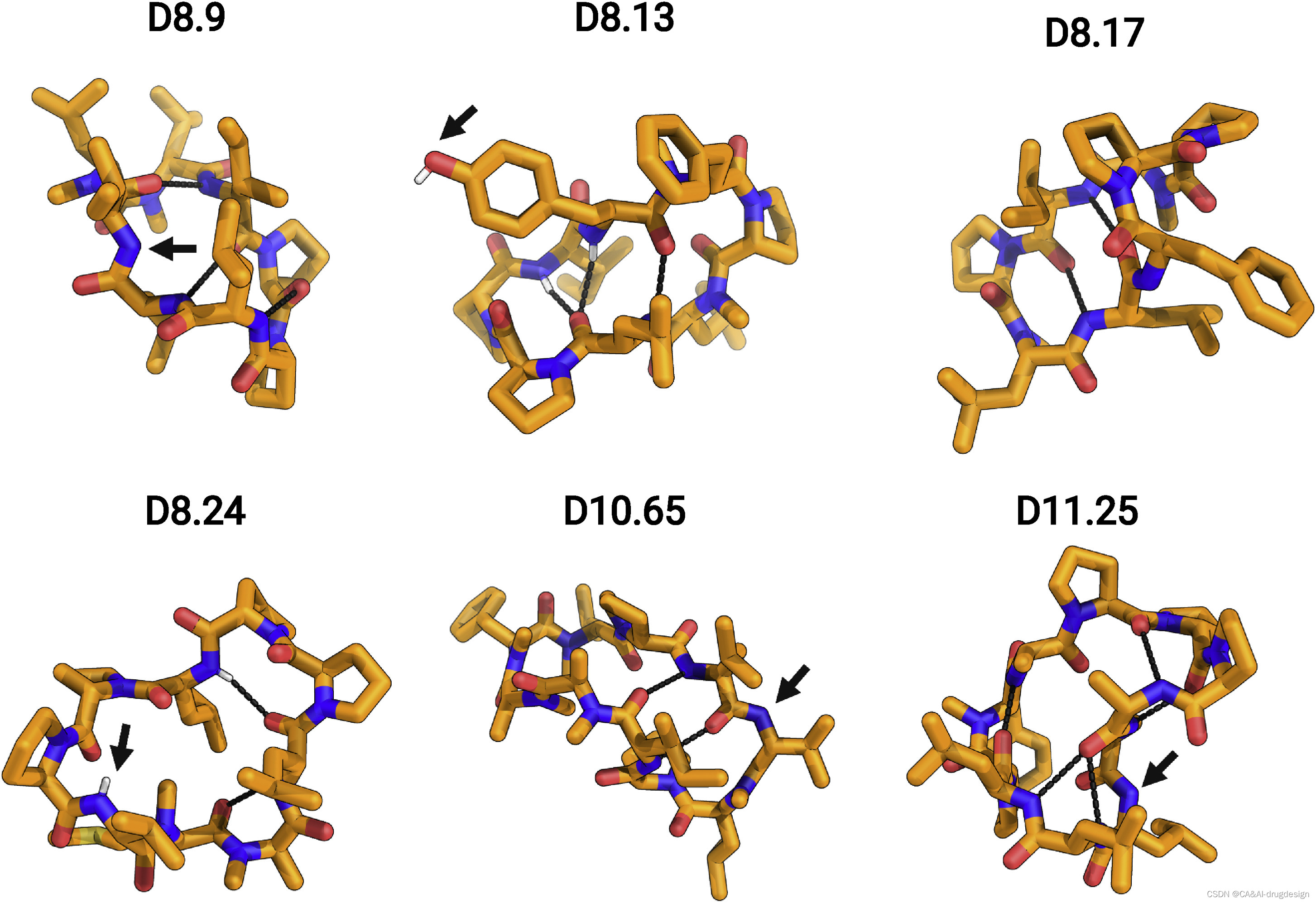
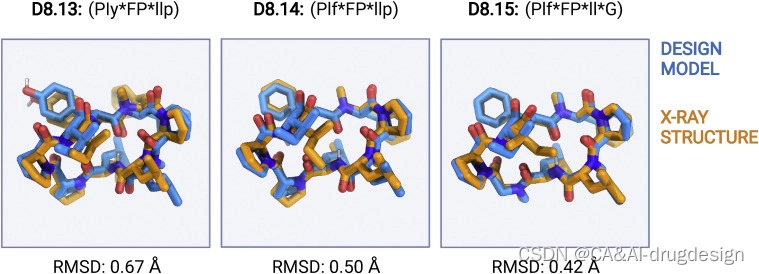
为了评估我们设计模型的结构准确性,并确认上述设计模型和穿膜性数据中存在的结构-活性关系,我们试图确定其实验结构。我们成功地将5个9-mers、6个10-mers和4个11-mers的高分辨率X射线晶体结构完全结晶化并解出。在这15个结构中,有3个9-mers、5个10-mers和4个11-mers大环肽与它们的设计模型密切匹配(骨架RMSD为1.2 A˚或更低)(图3;表S2)。
D9.8(RMSD 0.33 A˚)具有三个N-甲基化的氨基酸,结构由1个α turn,2个β turns和1个γ turn稳定。D10.31(RMSD 0.45 A˚)有两个N-甲基化的氨基酸和五个内部氢键来稳定大环结构;在异丙醇:水和乙酸乙酯:戊烷混合物中解析的结构是相同的。D10.1包含五个分子内氢键,晶体结构与设计模型几乎相同(骨架RMSD为0.27A˚,所有重原子RMSD为0.47A˚)。D10.21、D10.22和D10.23各含有5个N-甲基化氨基酸;D10.21由3个内部氢键和2个脯氨酸稳定,而D10.22和D10.23有2个内部氢键(骨架RMSDs分别为0.9、0.82和0.41 A˚)。D11.3和D11.4有五个内部氢键,它们的晶体结构与设计模型的骨架RMSD小于0.55 A˚。D11.1,设计模型和X射线晶体结构之间的骨架RMSD为0.43A˚,包含5个内部氢键,2个N-甲基化氨基酸和3个脯氨酸。
我们设计的大环肽的结构准确性加上大量的穿膜性测量,使我们对这些较大的肽的穿膜性和NH satisfaction之间的关系有了深入了解。**总的来说,几乎所有没有暴露NH基团的多肽都是可穿膜的。**与非设计的大环化合物的结果相反,穿膜性和大小之间没有强烈的相关性;事实上,一些10个和11个残基的肽是高度可穿膜的。不具有穿膜性或显示出低穿膜性的大环肽的晶体结构(图S3)进一步支持了NH satisfaction的重要性:这些结构与完全满足的大环肽设计模型不一致,这些结构包含了暴露的极性基团。尽管氢键satisfaction对于穿膜性似乎是必要的,但这并不充分:D10.21、D10.22和D10.23的X射线结构与它们的设计模型非常吻合(RMSD<1 A˚),没有unsatisfied的NH基团,但在transwell实验中没有穿膜性。
图S3 暴露和unsatisfied的极性基团对大环肽穿膜性的影响,与图1和图3有关
含有顺式肽键的大环肽的穿膜性
到目前为止,我们的结果确定了satisfying所有NH的计算设计大环肽可以稳定地产生远远超过Ro5限制的高穿膜性大环肽。我们接下来研究了对穿膜性的其他可能贡献。有人提出,顺式肽键可以提高穿膜性(Marelli et al., 2015)。 D8.31的X射线晶体结构在第3和第8位残基的两个N-甲基化的D-亮氨酸处有顺式肽键,这是由Nme-D-Pro(i+1)-Nme-D-aa(i+2) motif形成的罕见β-turn的一部分。D8.13包含三条脯氨酸,其中一条被设计为顺式肽键,由晶体结构中再现的芳香族AA(i+1)-脯氨酸(i+2) motif 稳定(Ganguly et al., 2012)。D8.6包含一排两个N-甲基化的氨基酸和四个分子内氢键;其中一个N-甲基化的D-丙氨酸在X射线晶体结构中经历了一个反式到顺式的转换,但由于转换发生在一个N-甲基化的氨基酸周围,大环肽中的整体NH satisfaction仍被保持。同样,在D8.9中,位于氨基酸第3位的N-甲基化D-亮氨酸经历了一个反式到顺式的转换,但保持了肽骨架的整体氢键satisfaction,D9.16的晶体结构也包含一个相对于设计模型的反式到顺式的omega翻转。
在我们的大环肽集合中,顺式肽键的存在与穿膜性的程度之间没有什么关联。含有顺式肽键的D8.9、D8.13、D8.14、D8.15、D8.6、D9.13和D10.62的穿膜性与具有相同残基数的全反式大环肽的穿膜性在同一范围。D8.13、D8.14和D8.15进一步说明了顺式肽键的次要贡献,它们的序列和结构相似(图S4),顺式肽键位于相同的位置;D8.13不具有穿膜性,D8.14和D8.15的PAMPA Papp为9.71×10-7和7.68×10-7 cm/s。D8.13有一个酪氨酸残基代替了苯丙氨酸残基,产生了一个unsatisfied的OH基团,可能阻止了穿膜性。
图S4 带有顺式肽键的大环肽的穿膜性差异,与图1有关
设计穿膜的变色龙(chameleonic)大环肽
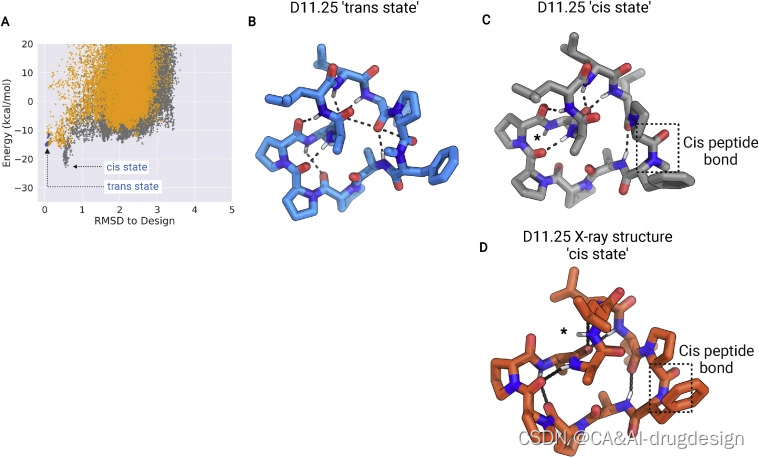
虽然上述结果表明顺式肽键本身并不增加穿膜性,但我们推断,肽键的顺反异构化可能是一个强大的设计原则,用于生成具有开放状态(极性基团暴露出来与治疗靶标相互作用)和穿膜封闭状态(所有NH基团形成肽内氢键)的肽(White et al., 2011)。顺反异构化的动力学相对较慢(几毫秒的时间尺度),这使得一个多肽可以填充多种构象,这些构象可以快速地相互转换,从而具有生物相关性,但可以通过核磁共振加以区分(Grathwohl and Wu¨ thrich, 1981)。有一些证据表明,环孢素中顺式-肽键周围的异构化会使不同的结合能力和穿膜状态相互转换(Wang et al., 2018)。为了测试这一假设,我们设计了进行顺反异构化的大环肽。D11.25为这一方向提供了一个起点:结构预测计算确定了两种非常相似的构象,它们的区别在于围绕序列中唯一的N-甲基化氨基酸的反式-顺式肽键翻转(图S5)。全反式构象由六个分子内氢键稳定,satisfy所有可用的NH基团,而顺式构象在晶体结构中得到了紧密的再现(骨架RMSD 0.53 A˚),在第10位暴露了D-亮氨酸的一个NH基团(图3;图S13**(没看到这张图,点开是table S1)**)。
图S5 肽键的顺反异构化产生了替代的低能量状态,与图3有关
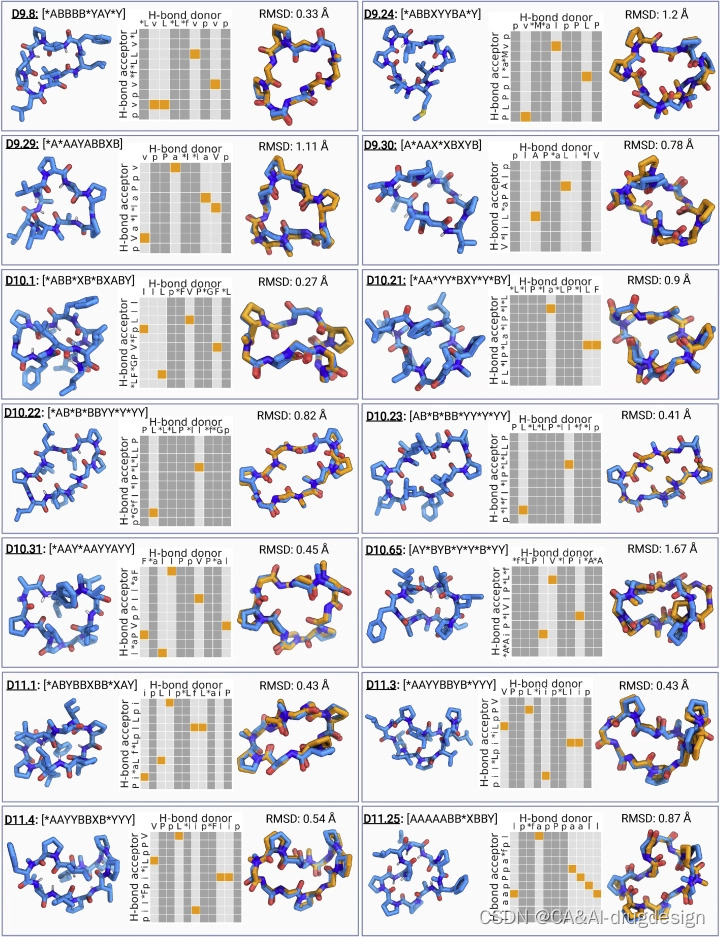
图3 9-12个氨基酸大环肽的计算设计和结构验证
我们着手系统地设计具有两个不同能量最小值的chameleonic多肽,这两个能量最小值因肽键周围的异构化而不同:一个是没有暴露的酰胺,可以穿越膜;另一个是将酰胺暴露在溶液中,因此有可能结合极性靶标位点。我们使用了三种方法来识别能够填充多种不同但几乎是等能状态的肽(Rosetta计算能量的差异小于5 kcal/mol)。首先,我们使用大规模的结构预测计算来生成许多大环肽的能量景观(Data file S1),搜索那些有两个或更多的最小值,并确定了45个大环肽,这些大环肽在两个状态之间的Rosetta计算能量差异小于5 kcal/mol。其次,对于另外20个计算出的状态之间的能量差异大于5 kcal/ mol的案例,我们开发了一个基于遗传算法的多状态设计方法(见STAR方法)来优化序列,使两个备选状态具有相似的能量。然后对这些新的序列进行了全能景观计算,以确认两个等能最小值的存在。第三,从晶体学确认的大环肽开始,我们引入了不稳定的突变,导致了能量景观中的第二个低能量最小值,并产生了另外两个肽,该肽被预测为采用两种状态。
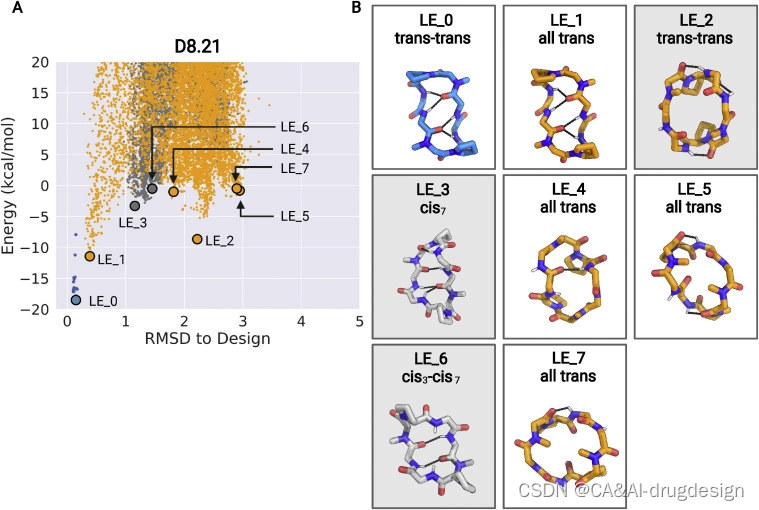
我们合成了67个大环肽,其中包括2个6-mers、2个7-mers、14个8-mers、25个9-mers、17个10-mers、5个11-mers和2个12-mers,预测它们具有可选择的低能量状态。其中,50个大环肽的PAMPA Papp大于1×10-7 cm/s,25个显示出明显的表观通透性(Papp>1×10-6 cm/s)(图2A;数据S3)。在Caco-2穿膜性试验中测试的10个大环肽中,有9个显示出明显的穿膜性:4个8-mers、2个9-mers、1个10-mers和2个11-mers显示Papp大于1×10-6cm/s。D8.21和D8.33显示Caco-2的Papp大于1×10-5cm/s(图2B)。我们选择了19种显示出明显穿膜性的多肽,用核磁共振进行进一步研究。这些大环肽的成功率较低,这与它们更大的极性、设计多种状态的挑战以及可能受构象异构化动力学限制的更慢的膜横向速率是一致的。在d6-DMSO和CDCl3中的1D 1H NMR表明,对于这些大环肽中的七个,结构化构象之间的平衡具有强烈的溶剂依赖性,在极性和非极性溶剂之间有构象状态或构象状态比率的切换。能量景观计算还显示,切换的大环肽通常导致两个以上的离散低能量状态–这是引入有利于一个以上状态的相互作用的预期结果。我们对每个大环肽的结构预测计算得出的低能组合进行了聚类,并根据该聚类中能量最低的结构给每个不同的最小状态(聚类)分配了一个标识符。对于用第一种方法设计的这七个大环肽中的三个(D8.21、D8.31和D9.16),我们成功地解出了与设计模型或预测的可替用的低能结构之一相匹配的晶体结构(图4)。然后我们使用2D NOESY、ROESY、TOCSY、13C-1H HSQC和13C-1H TOCSY-HSQC实验在CDCl3、d6-DMSO和50:50 d6- DMSO/H2O混合物中对这些大环肽进行了更详细的溶液NMR研究(见STAR方法),将每个构象中的脯氨酸和N-甲基化氨基酸分配为反式或顺式肽构象。异核NMR研究使用1H检测自然丰度的13C和15N核进行。然后对这些NMR数据进行分析,以确定每个肽的多种状态的结构,同时为三种肽在不同溶剂中提供总共11种溶液NMR结构(图4和S6;表S3和S4)。
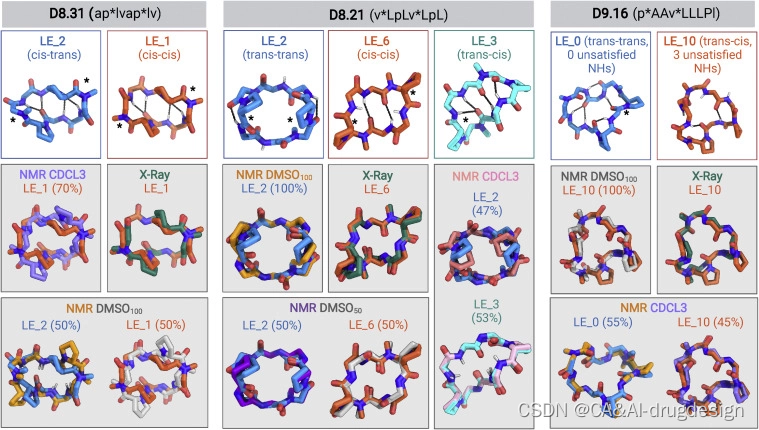
图4 构象转换大环肽的设计和结构表征
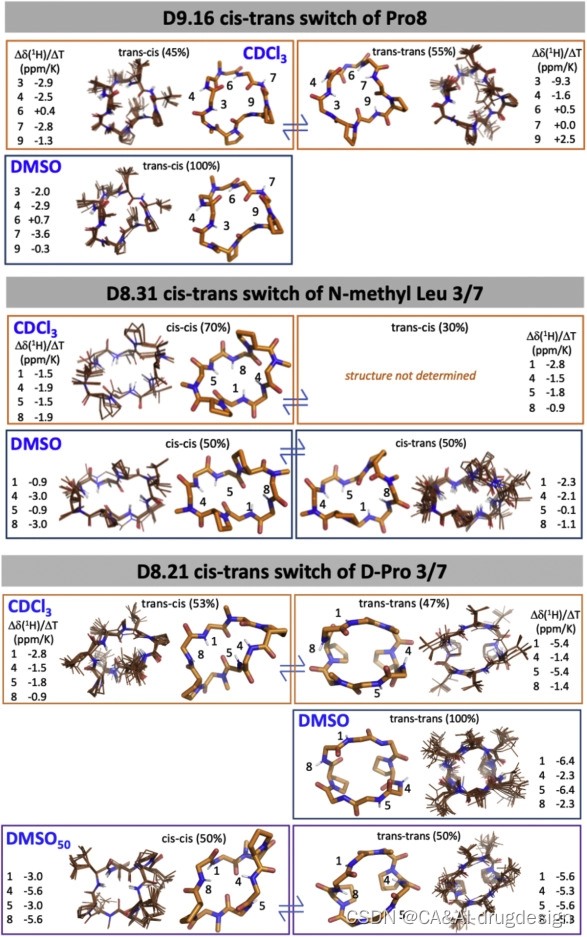
图S6 不同溶剂中的核磁共振结构,与图4有关
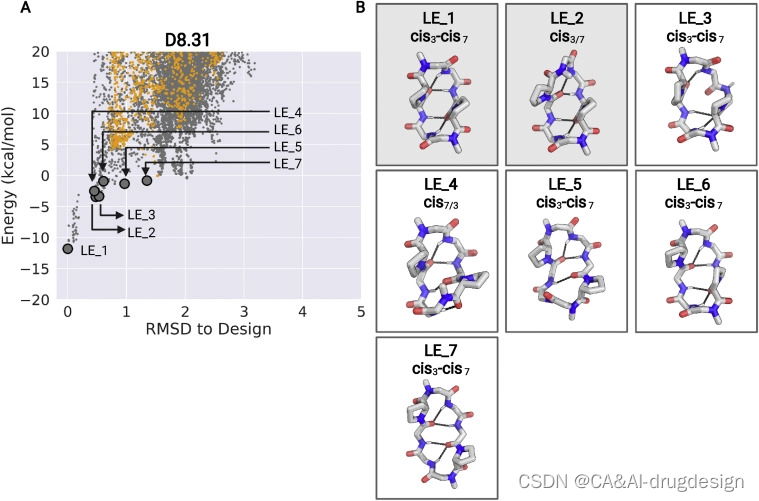
图S7 D8.31的低能量结构聚类,与图4有关
肽D8.31是一个具有对称重复序列的8个氨基酸大环肽(aplvaplv,*代表N-甲基化氨基酸)。最低能量状态(LE_1)是C2对称状态,两个N-甲基化氨基酸都处于顺式肽键构象(‘顺式-顺式’),第二低能量替代状态(LE_2)是不对称状态,一个N-甲基化亮氨酸处于顺式构象(‘顺式-反式’)(图4,左侧面板;图S6和S7)。顺反异构化发生在一个N-甲基化的氨基酸周围;因此,两种状态都没有unsatisfied的NH基团。乙酸乙酯:戊烷溶液中的晶体结构与顺式-顺式LE_1相似。在d6-DMSO溶液中,慢速交换中存在两种结构,顺式-顺式构象(~50%)与LE_1相似,顺式-反式构象(~50%)与LE_2相似(图4,左图)。在非极性CDCl3溶液中,也观察到两种构象,其中主要的形式(~70%)与LE_1状态相匹配(图4,左图)。这些实验性X射线晶体结构、溶液核磁共振结构和预测的D8.31的低能状态之间的对应关系表明,Rosetta计算可以指导采用多种状态的大环肽的设计。然而,由于两种不同的状态具有相同数量的暴露NH,这些数据并不能直接解决构象转换对穿膜性的贡献。更相关的是另外两个大环肽,D8.21和D9.16。
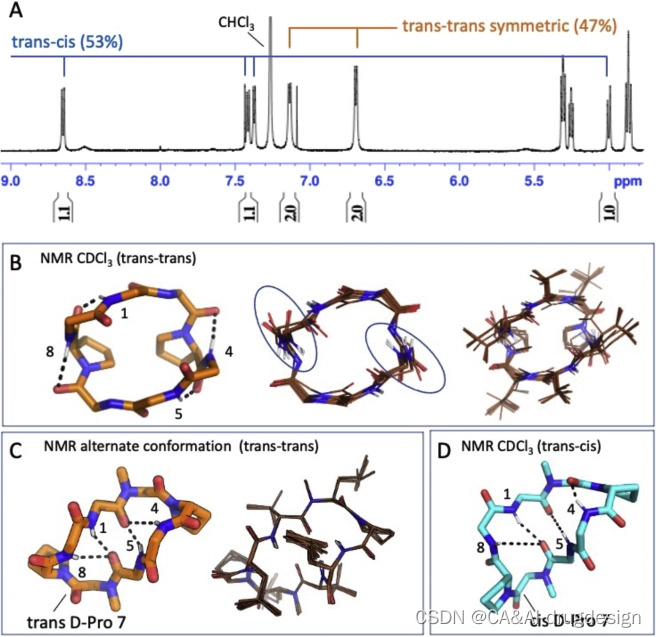
大环肽D8.21也有一个对称的重复序列(vLpLvLpL),预测有低能量的 "反-反 "状态(2个变体,LE_1和LE_2)、"反-顺 "状态(LE_3)和 “顺-顺”(LE_6)状态(图S8)。反式-反式的LE_2状态有暴露的NH基团,以及两个NH形成表面暴露的氢键。反式-顺式LE_3和顺式-顺式LE_6状态都有饱和的NH基团,在结构的核心部分形成氢键。水中条件下D8.21的X射线晶体结构是一个与LE_6类似的顺式-顺式构象(图4,中间部分)。在D6-DMSO溶液中,该肽是一个类似于LE_2的单一构象对称的反式-反式构象,而在50:50的D6-DMSO/2H2O中,它是这个相同的反式-反式构象(LE_2)和一个类似于X射线晶体结构和LE_6的顺式-顺式构象的50:50的平衡混合物(图4,中间部分)。在CDCl3中,该肽在平衡状态下也采用了两种构象状态;LE_2对称反式-反式构象,其数量为~47%(可能的替代状态见图S9C和图例),而不对称顺式-反式结构,其所有的NH都得到满足,与LE_3相匹配,其数量为~53%(图4,中间面板;图S7和S9)。**在这些核磁共振结构中观察到的NH基团的溶剂暴露程度也与酰胺化学位移数据的温度依赖性基本一致(图S6),与更多的极性溶剂相比,我们观察到在非极性CDCL3中所有NH都satisfied的反式-顺式LE_3构象体的明显稳定。**在更多的非极性溶剂中,具有内部氢键的NH基团的LE_3的稳定推动了构象转换,使这种状态的相对数量从极性溶剂中的0%增加到非极性溶剂中的53%,同时使unsatisfied 的表面NH基团的LE_2构象的数量从极性DMSO中的100%(以及50:50 d6-DMSO/H2O中的~50%)减少到CDCl3中的50%以下。
图S8 D8.21的低能量结构聚类,与图4有关
图S9 D8.21的核磁共振结构具有构象模糊性,与图4有关
D9.16有两个N-甲基化氨基酸和两个脯氨酸(pAAvLLLPl)。低能量设计模型是一个 "反-反 "构象(LE_0),没有unsatisfied的NH基团。预测的低能状态包括一个’‘反-顺’'状态(LE_10),有暴露的NH基团(图4,右面板;图S6和S10)。水中条件下的X射线晶体结构处于反式-顺式构象,与LE_10相匹配。在极性溶剂中(即50:50 d6-DMSO/2H2O和100% d6-DMSO),该肽具有单一的反式-顺式构象,有两个暴露的和一个埋藏的unsatisfied的NH基团,与LE_10相匹配(图4,右面板)。在非极性溶剂(CDCl3)中的核磁共振数据显示了两种状态之间的平衡:一种是与在d6-DMSO和晶体结构中观察到的LE_10状态相匹配的反式-顺式状态,另一种是与大环肽的反式-反式状态LE_0密切匹配的所有骨架NHs satisfied的反式-反式状态(图4,右面板)。在极性溶剂中,暴露NH基团的反式-顺式LE_10的数量从100%变为非极性溶剂中的45%,满足所有NH基团的反式-反式LE_0的数量从0%变为非极性溶剂中的55%。
总之,这些数据表明,D8.21、D8.31和D9.16大环肽确实存在多种状态,其中低能状态与实验中的晶体和核磁共振结构密切相关。与我们的构象转换设计策略一致,观察到的D8.21和D9.16的不同状态的相对数量与溶剂有关,在更多的非极性溶剂中,具有较少暴露或没有unsatisfied的NH基团的状态更受欢迎。总的来说,溶剂的变化改变了这些变色龙大环肽的低能量构象之间的平衡。按照设计意图,这些大环肽也具有穿膜性,但由于难以确定大环肽在穿膜过程中的状态,我们无法将这种穿膜能力具体归结为哪一个状态。值得注意的是,D8.21和D9.16在一种状态下暴露出骨架NHs,但仍保持着显著的穿膜性,这对未来设计可穿膜的大环肽来说是个好兆头。
口服生物利用度
口服生物利用度是一种值得做的治疗特性,它要求对胃肠道中的低pH值和蛋白酶具有稳定性,并能穿过肠道中的上皮细胞。我们选择了四种涵盖不同尺寸和体外穿膜率的大环肽,在啮齿动物模型中进行体内口服生物利用度和药动学研究。通过静脉注射(IV)、皮下注射(SQ)和口服(PO)途径单剂量给药后,测量了一种8-mer(D8.3.p1)、一种10-mer(D10.1)和两种11-mer肽(D11.2和D11.3)的血浆暴露。通过质谱法对血浆中的未修饰药物量进行量化,并以静脉给药为参考确定口服后血浆中未修饰药物的比例(%F)(见STAR方法)。在测试的剂量下,多肽的耐受性良好,没有任何不良反应。所有四种大环肽都有大量的口服暴露,与大多数其他天然口服吸收的多肽相比,显示出相当或更好的口服生物利用度。D8.3.p1、D11.2和D10.1具有7.5%和11%之间的良好%F(图5;数据S4)。11-mer设计大环肽,D11.3,在雄性瑞士白化病小鼠中进行了口服生物利用度测试,尽管其尺寸较大,但口服生物利用度(%F)非常高,为40%。这些设计的大环肽还显示了其他有利的类药属性,如长的血浆半衰期(T1/2)。D11.3在静脉给药后的T1/2为5.58小时,而D10.1在SQ给药后的T1/2为3.75小时(图5;数据S4)。总的来说,这些体内数据验证了这些经过计算设计和结构验证的多肽对低pH值和蛋白酶暴露是稳健的,并能有效地穿过肠道上皮屏障被吸收。
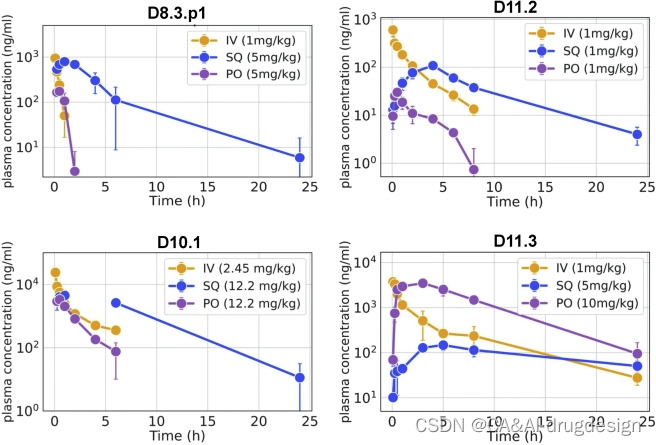
图5 设计的大环肽在啮齿动物模型中具有口服生物利用性
讨论
我们已经表明,精确控制结构的能力使我们能够稳定地设计出多种多样的穿膜大环肽,大大超出了以前发现的那些穿膜大环肽,主要是N-甲基化的β发夹(Bockus et al., 2015; Fouche´ et al., 2016; Hill et al., 2014; Ovadia et al., 2011; Wang et al., 2014)。与天然产物一样,我们设计的大环肽通过利用多种局部结构和内部氢键对极性基团进行构象屏蔽来实现这种高穿膜性。我们总共设计、合成和验证了84个结构不同的大环肽,它们具有良好到优秀的穿膜性,包括6-8个残基的高穿膜性和无N-甲基的大环肽,以及9-12个残基的穿膜大环肽,序列中只有一个N-甲基化氨基酸。PAMPA中大环肽的被动穿膜性转化为跨越Caco-2细胞的良好穿膜性和啮齿动物模型的口服生物利用度。穿膜的程度与实验结构和设计模型之间的sub-angstrom匹配度之间的强烈相关性突出了基于计算设计的结构控制的重要性:在我们成功确定晶体或核磁共振结构的35个大环肽中,25个与设计状态密切匹配(RMSD<1 A˚)的大环肽中,21个都是穿膜的(Papp>1×10-7 cm/s)。虽然35个大环肽中的29个的模型与相应的实验晶体或核磁共振结构非常接近(RMSD<1.2 A˚),表明设计方法具有非常高的准确性,但我们不能排除一种可能性,那就是缺乏X射线或核磁共振结构的大环肽折叠成对穿膜性很重要的替代构象。
本文介绍的设计方法和穿膜大环肽为开发口服生物可利用性的大环肽治疗药物提供了基础。精确控制结构的能力应能瞄准广泛的结合点的几何形状,而为穿膜性进行稳定设计的能力允许了靶向细胞内的靶标和口服给药。我们采用基于能量景观的方法来设计多肽,这些多肽表现出类似于环孢菌素的变色行为,随着溶剂极性的变化在亲脂状态和第二种相对极性的状态之间切换,应该能够设计出通过暴露的极性基团结合细胞内治疗靶标的、同时保持穿膜性的大环肽。
研究局限
虽然X射线晶体结构、核磁共振结构和穿膜数据之间有很好的匹配,但不能排除设计的大环肽有影响其穿膜性的替代构象,但没有结晶。虽然我们在这里重点讨论了通过肽内氢键实现穿膜,但侧链介导的unsatisfied的NH基团的屏蔽也可能是有效的;结构上验证的大环肽提供了模板,可以系统地评估NH屏蔽对穿膜性的影响。我们对chameleonic(变色)肽替代结构的控制还没有像对单态设计那样精确。随着能量函数精度的提高和景观取样的改进,这一点应该得到改善,包括方法上的改善和可用计算能力的提高。
未来的关键挑战是将靶标结合功能纳入大环肽,同时保持对结构和穿膜性的控制。**在本文中,设计的大环肽计算的重点是优化折叠精度和穿膜性。**设计结合剂可能需要加入极性或其他官能团,这些官能团与靶标进行能量上有利的相互作用,但对折叠或穿膜性(或两者)来说可能不是最佳的。我们目前的工作重点是通过研究极性侧链对穿膜性的影响来应对这一挑战,更广泛地说,是通过设计和表征各种细胞内靶标的结合物。
计算方法
对Rosetta软件套件的改进
为了在Rosetta软件套件中对含有N-甲基化氨基酸的环状肽进行设计、建模和结构预测,需要进行一些改进。首先,Rosetta的运动学机制被重新构建,随着聚合物内部自由度(如扭转角)的变化而跟踪更新原子的直角坐标。为了允许放置全部的化学基团(如N-甲基基团),我们改变了产生理想原子坐标的代码,其中原子坐标的位置依赖于肽键(如羰基氧和酰胺质子)。Rosetta的蛋白质切割点代码也进行了重构。这段代码确保Rosetta能量函数中的一个特殊项(称为断链chainbreak)在肽键位于运动树的断裂处时惩罚不良的肽键几何结构,这在环状肽中的某些点上不可避免地会发生;实际上,这使环状肽在能量最小化过程中不会打开。所需的修改使这个term与顺式肽键和N-甲基化兼容。
Rosetta的残基类型系统分为残基类型(独特的化学实体)和补丁(现有类型的小变体)。化学修饰,如质子化的N端、去质子化的C端或甲基化的侧链,通常用补丁来处理,它指示Rosetta通过改变现有残基类型的几何形状来增加一个变体,而不是增加一个全新的残基类型。我们以同样的方式支持骨架N-甲基化,添加一个补丁将一个氨基酸残基类型转换为其N-甲基化的等同物。然而,在骨架氮上增加一个甲基会大大改变一个给定的残基类型的骨架和侧链构象的偏好。我们为Rosetta的修补系统增加了支持,允许在修补过程中为被修补的残基类型指定新的主链电位和侧链旋转体库。然后,我们计算了N-甲基甘氨酸(肌氨酸)和N-甲基-L-丙氨酸的新主链Ramachandran电位,用AMBER GAFF力场计算了N-甲基甘氨酸,用高斯量子力学计算了N-甲基-L-丙氨酸的B3LYP/6-311+G(d,p)理论水平。由于第i个氨基酸残基上的N-甲基的立体斥力极大地影响了第i-1个氨基酸残基的构象偏好,这与脯氨酸在第i个位置时,脯氨酸侧链的立体斥力影响第i-1个残基的构象偏好的方式相同。Rosetta的rama_prepro能量项(计算主链电位,并对脯氨酸之前的残基应用不同的Ramachandran电位)被扩展到对N-甲基化位置之前的位置也应用前脯氨酸Ramachandran电位。对N-甲基甘氨酸和N-甲基L-丙氨酸也同样计算了前脯氨酸/前N-甲基Ramachandran电位。使用先前描述的MakeRotLib协议(Renfrew et al., 2012),生成了以下N-甲基化L-氨基酸残基的旋转体库:精氨酸、天冬酰胺、天冬氨酸、半胱氨酸、谷氨酸、谷氨酸、组氨酸、异亮氨酸、亮氨酸、赖氨酸、蛋氨酸、苯丙氨酸、丝氨酸、苏氨酸、色氨酸、酪氨酸和缬氨酸。Rosetta在对D-氨基酸残基构象取样或计算能量时,会自动反映L-氨基酸残基的主链电位和旋转体库,这一功能被扩展到N-甲基化氨基酸。在过去的工作基础上,我们将Rosetta的fa_dun侧链势概括为在计算侧链构象能量时使用MakeRotLib计算的旋转体概率(Mulligan et al., 2021),我们删除了以前包含的用于peptoid旋转体库的特例代码(Renfrew et al., 2012),允许所有旋转体采样和评分由新概括的旋转体代码处理。
在测试过程中,我们发现N-甲基、前面的羰基氧以及前面和相邻的侧链之间的密切相互作用导致了频繁的冲突,而这些冲突对N-甲基的旋转高度敏感。在某些情况下,这些冲突会导致Rosetta在旋转体优化过程中选择明显次优的侧链旋转体(打包)。因此,我们增加了对自由旋转的N-甲基的支持,使甲基氢原子的精确位置可以在包装或梯度白化能量最小化过程中得到调整。
为了促进设计和结构预测过程中的构象取样,我们修改了之前描述的广义运动学闭合(GeneralizedKIC)方法(Bhardwaj et al., 2016; Hosseinzadeh et al., 2017, Hosseinzadeh et al., 2021; Mulligan et al., 2021)。简而言之,基于GenerlizedKIC的大环肽构象取样是通过建立一个扩展链,随机化除6个骨干扭角外的所有扭角,然后分析解决剩余6个 "支点残基 "扭角的值,以确保断点处理想的肽键几何。为了更好地根据每个氨基酸的构象偏好进行偏向取样,考虑到其残基类型以及其骨架氮和下一个氨基酸的骨架氮上是否存在甲基,我们增加了一个新的GeneralizedKIC perturber,称为Randomize_backbone_by_rama_prepro,它使用与用于评分的相同的Ramachandran电位来偏重取样。由于枢轴残基扭转角是分析确定的,而不是从偏向分布中取样,所以一个给定的运动学封闭方案可能在Ramachandran空间的任何区域有骨架扭转值。因此,我们还增加了一个GeneralizedKIC过滤器,称为rama_prepro_check,它使用预先计算的Ramachandran电位来丢弃在Ramachandran空间的不良区域有枢轴残基的解决方案。由于运动学闭合计算和过滤是在自由度的原始向量上进行的,而不需要操作全原子分辨率模型,因此速度极快,可以在几秒钟内对成千上万的构象进行采样;设计协议中的下游设计步骤占据了大部分的计算时间。GeneralizedKIC渗透器和过滤器的文档可以在Rosetta帮助维基上找到:https://www.rosettacommons.org/docs/latest/scripting_documentation/RosettaScripts/composite_protocols/generalized_kic/GeneralizedKICperturber 和 https://www.rosettacommons.org/docs/latest/scripting_documentation/RosettaScripts/composite_protocols/generalized_kic/GeneralizedKICfilter。
此外,还开发了一个新的残基选择器,称为Unsat,允许识别具有unsatisfied的骨架氢键供体或受体的氨基酸残基,以便在RosettaScripts或PyRosetta设计协议中允许自动进行N-甲基化。这个选择器与不对称和对称结构都兼容。Unsat残基选择器的文档可以在Rosetta在线文档(https://www.rosettacommons.org/docs/latest/scripting_documentation/RosettaScripts/ResidueSelectors/ResidueSelectors#residueselectors_conformation-dependent-residue-selectors_unsatselector)中找到。
我们在Rosetta的simple_cycpep_predict程序中增加了对使用N-甲基化氨基酸的支持,该程序用于从氨基酸序列预测大环肽结构,并估计池中每个设计的多肽的折叠倾向,以便对设计的大环肽进行排序和优先排序,进而进行合成。在肽构象取样过程中,simple_cycpep_predict使用上述的GeneralizedKIC方法对大环肽的封闭构象进行快速取样,使用新实施的 randomize_backbone_by_rama_prepro GeneralizedKIC perturber来偏置取样,使用rama_prepro_check GeneralizedKIC filter来丢弃在枢轴原子上有不良骨架构象的方案。Rosetta的基于能量的聚类应用也进行了修改,使其能够聚类N-甲基化的肽,并将骨架甲基的碳作为对齐结构和计算RMSD值时的原子。这些应用程序的文档可以在Rosetta帮助维基上找到,分别是https://www.rosettacommons.org/docs/latest/structure_prediction/simple_cycpep_predict#full-inputs_additional-flags-for-n-methylated-amino-acids 和 https://www.rosettacommons.org/docs/latest/application_documentation/analysis/energy_based_clustering_application。对这些变化的测试发现了一个与Rosetta能量函数中Lazaridus-Karplus项中的一个未初始化的变量有关的小错误,这个问题也得到了纠正。
截至2021年10月(Rosetta Git修订版a9ab4ac590fd0a4e2def5739deaeab02c72c949d),这些改进都已纳入Rosetta的公开版本,但N-甲基化旋转体库除外。由于它们的大小,这些库没有与Rosetta一起发布(但N-甲基色氨酸旋转体库除外,用于单元测试),但都与非经典氨基酸库捆绑在一起单独发布。还增加了广泛的单元测试,以确保涉及N-甲基化氨基酸的能量计算不受循环变异或镜像的影响。Rosetta的源代码和编译后的二进制文件可以免费提供给学术界、政府和非营利性的用户,也可以从华盛顿大学获得许可,用于企业和营利性的使用。要下载和使用Rosetta,请访问https://www.rosettacommons.org/software/license-and-download。
结构化穿膜肽的计算设计
我们修改了之前描述的基于Rosetta广义运动学闭合(GeneralizedKIC)的大环肽设计协议(Bhardwaj et al., 2016; Hosseinzadeh et al., 2017, Hosseinzadeh et al., 2021; Mulligan et al., 2021),以便能够设计能够穿越脂质膜和细胞屏障的6到12个氨基酸大环肽。大环肽设计的所有步骤都在RosettaScripts(Fleishman et al., 2011)中实现。
简而言之,我们选择了一个大环肽的尺寸,并通过使用Rosetta PeptideStubMover(Bhardwaj et al., 2016)构建所选氨基酸长度的线性聚甘氨酸骨架来启动设计计算。在这项工作中,我们对6到12个氨基酸之间的每个大环肽尺寸进行了单独的设计运行。接下来,我们在聚甘氨酸肽的最后一个残基的 "C "原子和第一个残基的 "N "原子之间声明了一个键,并使用Rosetta PeptideCyclizeMover(Hosseinzadeh et al., 2017)设置了N-C末端环化的距离、角度和二面体约束。我们随机选择一个残基作为 “锚定残基”,另外三个残基作为 “支点残基”。所有残基的omega扭转被设置为180°;锚定残基的ɸ和ψ扭转是使用SetTorsion mover(Bhardwaj et al., 2016)从平底镜面对称的Ramachandran表中随机选择的。随后,我们使用GeneralizedKIC移动器从线性肽中识别环状聚甘氨酸肽(Bhardwaj et al., 2016)。在GenKIC移动器内,非枢轴和非锚定残基的ɸ和ψ二面体从平底镜面对称的Ramachandran表中随机抽取。
枢轴残基的ɸ和ψ二面体是通过运动学封闭算法(Bhardwaj et al., 2016; Coutsias et al., 2004, Mandell et al., 2009)分析计算的,以找到能得到N-C环状肽骨的二面体角的组合。闭合的标准被进一步定义为包括最低数量的内部骨架对骨架的氢键。所需的内部氢键数量基于大环肽的长度:6-7个氨基酸需要至少1个内部氢键,8-9个氨基酸需要2个氢键,10个或以上氨基酸的大环肽需要3个内部氢键。在GenKIC返回多个环状解决方案的情况下,我们根据自定义的Rosetta能量函数选择能量最低的解决方案,该函数仅包括fa_rep、fa_atr、hbond_sr_bb、hbond_lr_bb、rama_prepro和p_aa_pp得分项(Alford et al., 2017)。对于每个成功封闭的环状骨架,我们使用Rosetta FastDesign移动器设计一个氨基酸序列,试图使大环肽的整体能量最小化(Bhardwaj et al., 2016)。
由于我们的目标是设计能够被动穿越脂质膜的大环肽,我们通过将具有这种 ‘unsatisfied’的骨架NH基团的氨基酸突变为N-甲基化的氨基酸,从而去除不参与氢键的NH基团。使用Unsat选择器选择具有unsatisfied的NH基团的残基,并使用ModifyVariantType移动器将其突变为N-甲基化版本。然而,鉴于N-甲基化氨基酸的不同扭转偏好,对N-甲基氨基酸的突变可能也会暴露出其他氨基酸的NH基团。因此,我们采用了迭代的方法,进行了三轮的氨基酸序列设计,并在中间对暴露的骨架NH基团进行了N-甲基化。在第一轮设计中,我们使用了一个带有加权(5倍)骨架氢键得分项的能量函数,以支持更多的内部backbone-to-backbone的氢键。
在第二轮设计中,我们使用了标准的Rosetta beta_nov16权重与约束条件(Alford et al., 2017; Park et al., 2016)。在第三轮设计中,我们也允许卡氏最小化。在设计过程中,我们只允许疏水性氨基酸,只允许D型氨基酸出现在有正ɸ的残基位置,只允许L型氨基酸出现在有负ɸ值的残基位置。对于一些运行,我们还使用了AddCompositionConstraintMover来限制所设计的肽中允许的脯氨酸、D-脯氨酸和一些笨重的疏水氨基酸的最小和最大数量(Hosseinzadeh et al., 2017)。鉴于合成具有多个N-甲基化氨基酸的肽的难度,我们使用Rosetta SimpleMetrics(Adolf-Bryfogle et al., 2021)根据设计模型中N-甲基化氨基酸的总数和最终设计状态中缺乏任何暴露的NH基团来过滤设计模型。
对于每个选定的尺寸范围,大约有105个大环肽被抽样。如前所述(Hosseinzadeh et al., 2017),设计模型根据从骨干二面角计算出的扭转仓串进行聚类。接下来,我们使用Rosetta simple_cycpep_predict应用程序(Bhardwaj et al., 2016; Hosseinzadeh et al., 2017)对来自不同聚类的最低能量得分设计进行结构预测,如前所述(Hosseinzadeh et al., 2017)。我们评估了来自结构预测计算的能量与RMSD-to-Design图(图S2),并根据低能量状态的数量选择了结构化和构象转换的肽。在结构预测计算确定的构象比设计模型的能量低(用_LE表示)的情况下,低能量的构象被用作结构比较的模型。
Rosetta大分子建模套件可从 https://rosettacommons.org/software 下载。描述上述协议中使用的RosettaScripts和组件细节的Rosetta文档可在https://new.rosettacommons.org/docs/latest/Home。在Data S5中还可以找到一个包含脚本和命令行标志示例的文件夹。下面提供一个设计带有N-甲基化氨基酸的10个氨基酸多肽的RosettaScripts案例。
构象转换肽的多状态设计
我们使用多状态设计方法来生成变色龙大环肽。具体来说,我们在PyRosetta-3中实现了一个遗传算法,优化突变以获得所设计的氨基酸序列的两个等能低能量状态(脚本和所需文件见数据S5)。起始序列被用来生成1000个变体的列表,这些变体包含对不同疏水残基的突变,同时保持原有的手性和N-甲基化模式。然后将每个突变体序列穿到原始骨架构象上,并使用REF2015 Rosetta能量函数进行评分(Park et al., 2016)。序列被过滤,以确保两个骨架上的序列的总能量小于10kcal/mol,并且两个状态之间的差异小于6kcal/mol。为了选择在保持低能量的同时稳定两种构象的序列,然后给序列一个最终的分数,等于(-5×abs(eA-eB))-(eA-eB),其中eA和eB是一个给定的序列分别穿在第一和第二构象上的Rosetta分数。基于这个指标的最好的500个序列被带入下一个评估周期,每个序列的一个点突变体被添加到列表中,然后重复这个过程。这个算法运行了1000代,然后选择最佳序列。进行结构预测,以确保两种期望的构象状态都作为构象景观中的低能最小值。
结构化多肽向构象切换多肽的转化
对于一些晶体学确认的大环肽,我们试图找出可以创造二级等能点的氨基酸替换。我们实现了一个PyRosetta脚本,该脚本通过给定结构的每个氨基酸位置进行循环,并在保持手性和N-甲基化模式的同时将原始残基突变为其他疏水氨基酸(脚本和所需文件见补充资料)。然后使用Rosetta FastRelax协议(Bhardwaj et al., 2016)对原始结构的每个突变版本进行能量最小化,以缓解突变所引起的任何应变。使用Rosetta cycpep_predict应用程序(Bhardwaj et al., 2016; Hosseinzadeh et al., 2017)评估了全套突变序列-结构对的结构-能量景观,并选择了由顺/反异构化分开的、具有等能量交替状态的大环肽,用于实验表征。
对设计的大环肽进行结构预测
我们使用之前描述的Rosetta simple_cycpep_predict应用程序(Bhardwaj et al., 2016; Hosseinzadeh et al., 2017)来评估设计的大环肽的氨基酸序列,并对其进行构象景观。我们使用Rosetta@Home平台进行结构预测计算。对于每个大环肽,我们产生了>104个设计的氨基酸序列的能量最小化的循环构象。对于每个构象,我们使用Rosetta REF2015能量函数计算了对比设计模型的RMSD以及能量(Park et al., 2016)。在Rosetta@Home上运行脚本所使用的参数相当于在计算集群上运行以下脚本。
/home/gauravb/cycpep_cst_fix_copy/Rosetta/main/source/bin/simple_cycpep_predict.default.linuxgccrelease -in:path:数据库 /home/gauravb/cycpep_cst_fix_copy/Rosetta/main/数据库 @input.flags -nstruct 100
下面是一个flags文件(input.flags)的例子。
数据和代码的可用性
所有用于设计和验证肽的脚本都在Rosetta大分子建模套件和PyRosetta中实现。Rosetta软件套件和PyRosetta可以从https://www.rosettacommons.org/ 和https://www.pyrosetta.org/。用于设计大环肽的脚本和命令行标志的例子包括在补充资料文件中。

















 3099
3099

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









