







分子动力学(MD)的一般指南:
(1)选择分子动力学软件:
选择适合您研究需要的分子动力学软件包。常用的软件包括 GROMACS、AMBER、NAMD、CHARMM 和 LAMMPS。
(2)准备分子结构:
获取相关分子的原子坐标和拓扑文件。具体方法如下:使用分子建模工具(如 Avogadro、PyMOL 或 VMD)从头开始创建分子结构。从公共数据库(如蛋白质数据库、PubChem)或以前的模拟中检索分子结构。如果没有现成的配体或小分子,则构建配体或小分子。
(3)虚拟体系的搭建
模拟体系是被高度简化的,一般包括水、无机离子、蛋白质、(小分子)。
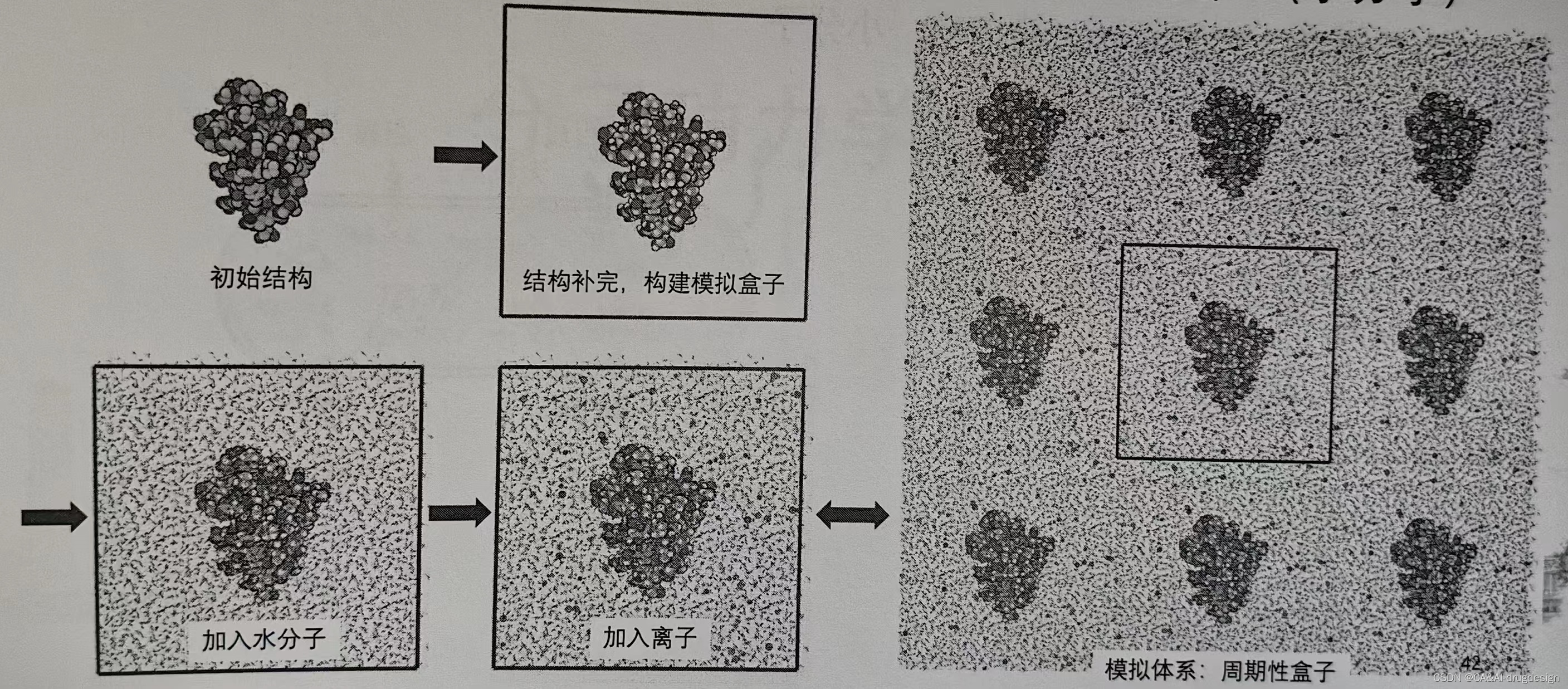
虚拟体系搭建的原则主要有三点:
一、希望模拟体系始终保持在水溶液中
不包含试管壁等其他因素
二、使用周期性边界条件(PBC)能够使体系尽可能地小
中心盒子中心粒子在运动时,该粒子的镜像在镜像盒子中以完全相同的方式运动。
当粒子跨过中心盒子的周期性边界,进入镜像盒子,它会被视作从反方向进入中心盒子。
水分子能够始终包围蛋白质。
三、周期性盒子可以有不同的形状
立方体最稳妥
截顶八面体可以明显节省计算量
虚拟体系搭建的具体流程如下:
1、定义系统:
结合目标分子、溶剂、离子和其他相关分子的分子结构,创建完整的系统。根据需要添加或移除离子,确保体系呈电中性。
2、溶解体系:
用近似周围环境行为的溶剂模型(如水、有机溶剂或脂质双分子层)包围分子成分。
选择合适的溶剂模型,并确保溶质分子周围有足够的缓冲溶液。
3、添加离子:
如果您的系统需要离子来保持电荷中性或模拟特定的离子条件(如盐浓度),则应添加离子并将其适当置于溶剂中。
4、能量最小化:
执行初始能量最小化以放松系统并消除原子间的不利接触和碰撞。这通常使用最小化算法(如最陡下降算法或共轭梯度算法)完成。
(4)虚拟环境条件
虽然分子动力学模拟可以直接限定粒子数目、固定体积和维持能量来进行恒容恒能量系综的模拟,但实验条件一般都是在室温或者体温以及标准大气压下进行。因此在实现分子动力学模拟时还需要考虑温度和压强的耦合。
常用的温度耦合方法
·Berendsen弱耦合方法、Andersen恒温器法、Nos-Hoover方法和Velocity-rescaling方法
常用的压强耦合方法
·Berendsen弱耦合方法,Parrinello-Rahman方法和Martyna-Tuckerman-Tobias-Klein(MTTK)方法
温度和压强耦合方法的两种用途
·体系预平衡
把模拟体系的温度和压强调整到期望值附近,并且去除体系中的不合理的局部结构
·正式采样
收集数据,用于计算体系的各种宏观和微观性质
希望产生的结构能够符合等温等压系统
推荐使用的温度和压强耦合方法
| 温度耦合算法 | 压强耦合算法 | |
|---|---|---|
| 预平衡 | V-rescale或Berendsen | Berendsen |
| 正式采样 | V-rescale或Nose-Hoover | Parrinello-Rahman或Berendsen |
(5)分子动力学模拟-模拟原子间的相互作用
具体流程如下:
1、平衡系统:
进行平衡运行,为生产 MD 模拟准备系统。这包括将系统加热至所需温度。
根据需要进行压力控制(如 NPT 或 NVT 组合)。
运行短时间 MD 模拟,让系统适应所需的条件。
2、MD 模拟:
在所需的集合(NPT 或 NVT)和条件(温度、压力和时间步长)下启动MD 模拟。
在模拟过程中监控并记录轨迹数据,以便进行后续分析。
3、分析和后处理:
分析 MD 轨迹数据以提取相关信息,如结构变化、热力学性质和分子相互作用。
使用分子可视化工具(如 VMD、PyMOL)和数据分析软件对模拟结果进行可视化和解释。
理论知识
1、模拟原子之间的相互作用需要用到分子力场,分子力场U包括两大部分:成键相互作用和非键相互作用
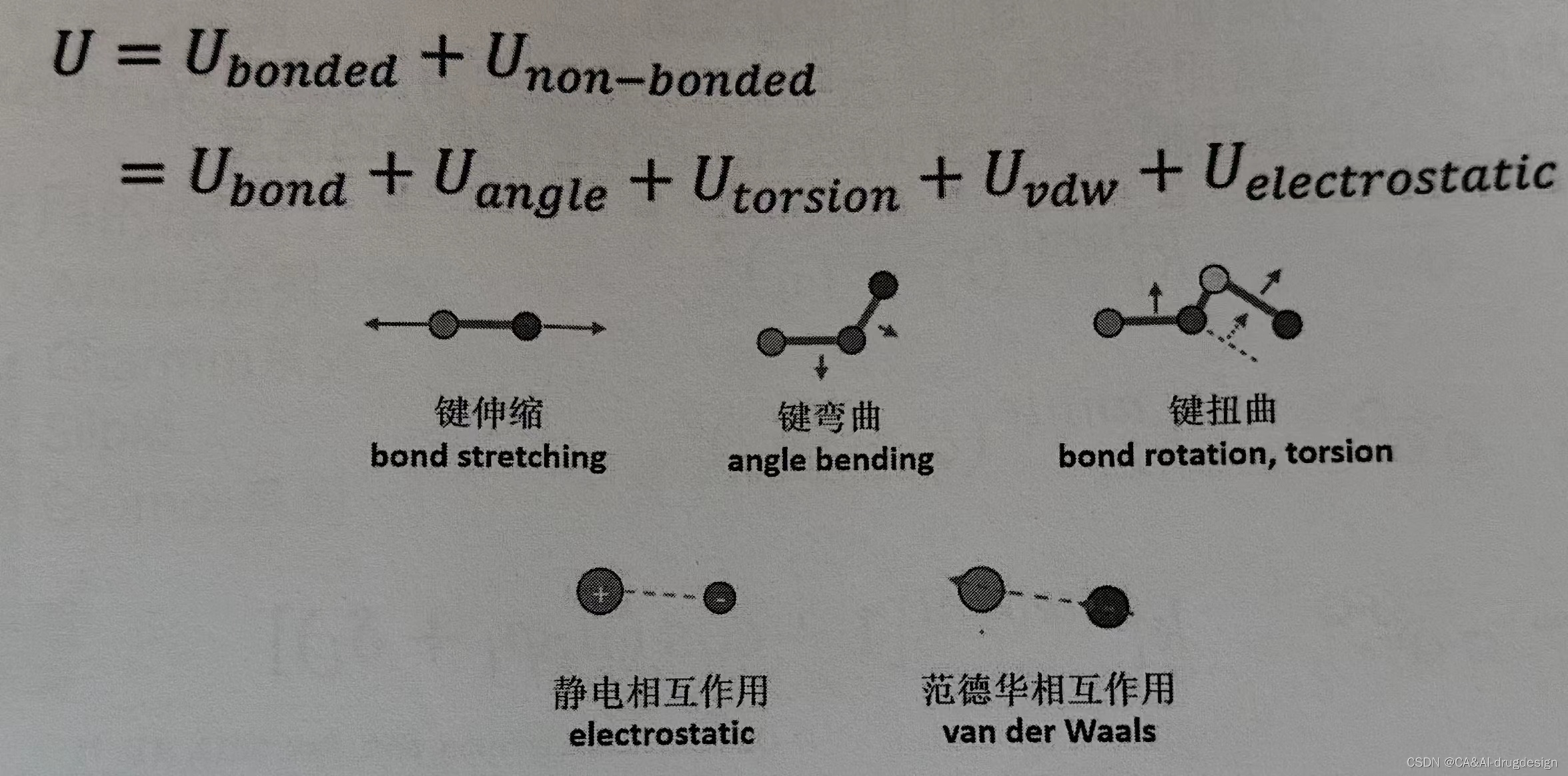

不要用口碑很差的力场
一些较为常用和较新的力场
注意:蛋白质力场,小分子力场,和显式水分子模型的搭配
| 系列 | 推荐版本 | 小分子 | 水分子 |
|---|---|---|---|
| AMBER | 99sb-ILDN,14sb | GAFF, GAFF2 | TIP3P |
| 19sb | OPC | ||
| CHARMM | 36m | CGenFF | TIP3P |
| OPLS | 3e | 3e | SPC |
| GROMOS | 53a6,54a7, 54a8 | 2016H66 | SPC |
小分子力场的生成
| 软件名称 | 力场名称 | 网址 |
|---|---|---|
| Antechamber | GAFF | ambermd.org/tutorials/basic/tutorial4b/index.htm |
| Acpype | GAFF | github.com/alanwilter/acpype |
| ATB | GROMOS | atb.uq.edu.au/index.py |
| CGenFF | CGenFF | cgenff.umaryland.edu |
| LigParGen | OPLS | zarbi.chem.yale.edu/ligpargen/index.html |
| PolyParGen | OPLS | polypargen.com |
| SwissParam | MMFF | www.swissparam.ch |
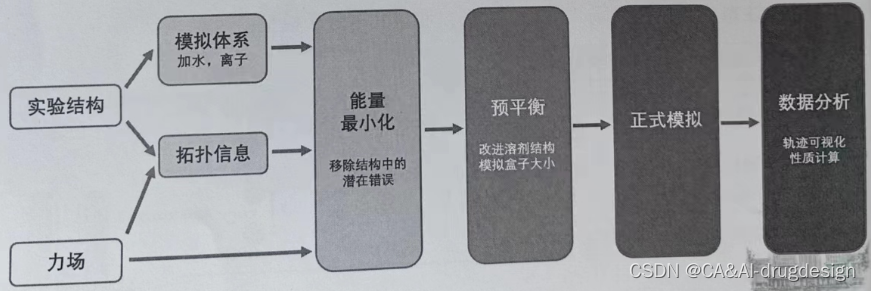
2、分子动力学工作流程

















 1029
1029

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









