







TPM是什么?为什么要计算TPM
我们通常所说的TPM,RPKM,FPKM,其实是三种对测序的Row reads count进行归一化的手段。
TPM: Transcripts Per Kilobase of exon model per Million mapped reads (每千个碱基的转录每百万映射读取的Transcripts)。
除了TPM还有RPKM和FPKM,不过科研人员主要使用 TPM的方法对基因表达量进行归一化。
RPKM: Reads per kilobase of transcript per Million reads mapped(每千个碱基的转录每百万映射读取的reads)
FPKM: Fragments Per kilobase of transcript per Million reads mapped(每千个碱基的转录每百万映射读取的fragments)
为什么不能直接使用Row reads count呢?
在RNA-seq中,每个mRNA转录本的表达水平是由 映射(mapped) 片段的总数来衡量的,这预计与其丰度水平成正比。然而,在计算Row reads count 之后,数据归一化对于确保准确推断基因表达是必不可少的。
mapped 到给定基因的 Row reads count 在样本或条件之间不具有可比性,因为测序深度或文库大小(mapped reads 的总数) 通常因样本而异。
一个样本中不同基因的Row reads count也不能直接进行比较,因为与表达水平相似的较短转录本 (transcript) 相比,较长的转录本具有更多的读数。
因此,为了消除测序数据中的技术偏差,需要使用归一化,而不是直接使用Row reads count,例如RPKM(每千碱基每百万次映射的转录的读数)、FPKM(每百万个片段的每千基的转录的片段)和TPM(每百万条的转录)。
FPKM与RPKM密切相关,但用片段(Pair reads) 取代了单端测序(这种命名的原因是历史的,因为最初的读取是单端的,但随着 pair-end 测序的出现,现在谈论片段更有意义,因此也就是FPKM).
RPKM、FPKM 和 TPM这三个指标试图标准化测序深度和基因长度。
目前TPM的计算方式更加科学,被研究人员普遍认可。
计算公式:
# 1.设置工作路径
setwd("D:/filename/")
# 2. 读取数据 【数据格式要保存为csv格式】
mycounts<-read.csv("010_gene_RRC_Guy11.csv")
head(mycounts)
rownames(mycounts)<-mycounts[,1]
mycounts<-mycounts[,-1]
head(mycounts)
kb <- mycounts$Length / 1000
head(kb)
# 3. 计算TPM 【至少要有两列Row reads count数据,才能计算,否则报错。其中的mycounts[,1:2]根据你的样品数量,像这里我只有两个样品就是1:2,如果你是10个样品就是 1:10】
countdata <- mycounts[,1:2]
head(countdata)
rpk <- countdata / kb
head(rpk)
tpm <- t(t(rpk)/colSums(rpk) * 1000000)
head(tpm)
# 4.保存结果
write.table(tpm,file="011_gene_Guy11_TPM.tsv",sep="\t",quote=F)
输入文件格式:
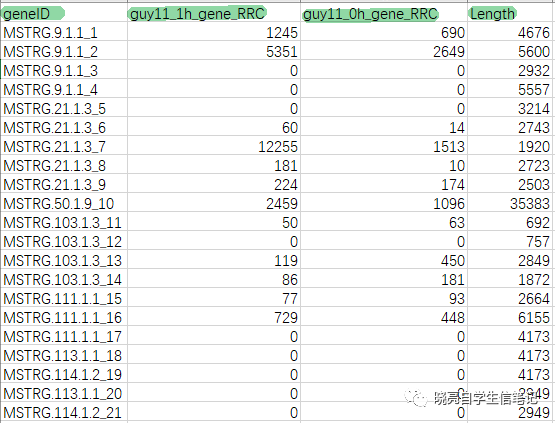
- 输入的文件 010_gene_RRC_Guy11.csv 具体数据格式,四行,第一行为基因id,第二行-第n行为你的基因表达量(Row reads count),第n+1行为你的基因Length,最后保存在csv格式,如下图。
TPM 与 DESeq2 或 TMM 的归一化结果还是有一些差异,目前来看 DESeq2 归一化方法对于大多数情况,结果更加准确。
因此,重要的是在选择归一化方法时考虑自己的实验目的和实验材料,而不是任意地将单一方法用于所有实验数据。
研究人员需要意识到各种方法所做的假设,以及可能违反这些假设的数据特征,以便为他们的研究选择正确的归一化方法。
参考文献
-
Zhao S, Ye Z, Stanton R. Misuse of RPKM or TPM normalization when comparing across samples and sequencing protocols. RNA. 2020 Aug;26(8):903-909.
-
Zhao, Y., Li, MC., Konaté, M.M. et al. TPM, FPKM, or Normalized Counts? A Comparative Study of Quantification Measures for the Analysis of RNA-seq Data from the NCI Patient-Derived Models Repository. J Transl Med 19, 269 (2021).
您可能还想看:
**搜文献神器,谷歌搜索,谷歌学术免费用
**















 884
884











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









