







ICLR 2020 | GraphAF: a Flow-based Autoregressive Model for Molecular Graph Generation

Paper: https://arxiv.org/abs/2001.09382
Code: https://github.com/DeepGraphLearning/GraphAF
graphhaf:一个基于流的分子图生成自回归模型
这篇是来自北京大学和上海交通大学的Chence Shi等人在2020年的ICLR上发表的会议论文。
摘要
分子图谱的生成是药物发现的一个基本问题,越来越受到人们的关注。这是一个具有挑战性的问题,因为它不仅需要生成化学上有效的分子结构,同时也需要优化它们的化学性质。受深度生成模型的启发,本文提出了一种基于流的自回归图生成模型,称为GraphAF。GraphAF结合了自回归和基于流的方法的优点:(1)数据密度估计的高模型灵活性;(2)用于训练的高效并行计算。(3)一个迭代采样过程,它允许利用化学领域的知识进行价键检查。实验结果表明,即使没有化学知识规则,GraphAF也能生成68%的化学有效分子,而有化学规则则能生成100%的有效分子。GraphAF的训练过程比现有的最先进的方法GCPN快两倍。通过强化学习对目标导向性能优化模型进行微调后,GraphAF在化学性能优化和约束性能优化方面都取得了最先进的性能。
介绍
设计具有理想性能的新型分子结构是药物发现和材料科学等各种应用中的一个基本问题。这个问题非常具有挑战性,因为化学空间本质上是离散的,而整个搜索空间是巨大的,据信高达 1 0 33 10^{33} 1033。机器学习技术在分子设计领域看到了巨大的机会,这要归功于这些领域的大量数据。近年来,越来越多的人致力于开发能够自动生成化学上有效的分子结构并同时优化其性质的机器学习算法。
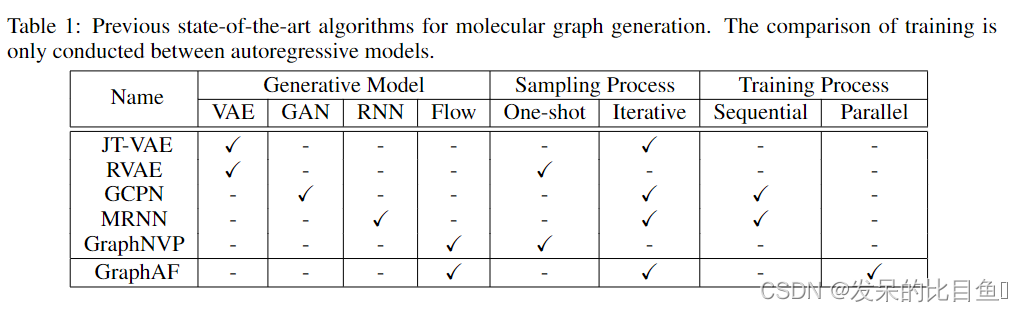
具体来说,通过将分子结构表示为图,并使用深层生成模型生成图结构,例如变分自动编码器(VAEs)、生成对抗网络(GANs)和自回归模型,已经取得了重大进展。例如,Jin提出了一种用于分子结构编码和解码的连接树VAE (joint Tree VAE)。De Cao和Kipf研究了如何使用GAN生成分子图。You提出了一种称为图卷积策略网络(GCPN)的方法,该方法将分子图生成形成一个序列决策过程,并基于现有的图子结构动态生成节点和边。他们使用强化学习来优化生成图结构的性质。最近,另一项非常相关的工作molecular arrnn (MRNN)提出使用自回归模型生成分子图。基于自回归的方法,包括GCPN和MRNN,在分子图生成的各种任务中表现出非常有竞争力的性能。
最近,除了上述三种生成模型外,规范化流程也取得了显著的进展,并已成功地应用于各种任务,包括密度估计、变分推断和图像生成。基于流的方法定义了潜在基分布(如高斯分布)和真实的高维数据(如图像和语音)之间的可逆转换。这种可逆映射允许计算准确的数据可能性。同时,通过隐藏空间与观测空间之间的多层非线性变换,流具有较高的数据密度建模能力。此外,根据不同应用的需求,可以设计不同的架构来促进快速训练或快速采样。
受自回归模型的现有研究成果和具有标准化流程的深层生成模型的最新进展的启发,我们提出了一个基于流程的自回归模型,称为GraphAF,用于分子图生成。GraphAF有效地结合了自回归和基于流的方法的优点。它有很高的模型容量,因此能够模拟真实世界分子数据的密度。GraphAF采样过程设计为自回归模型,根据已有子图结构动态生成节点和边缘。与GCPN和MRNN等现有模型相似,这种连续生成过程允许在每个生成步骤中利用化学域知识和价键检查,从而保证生成的分子结构的有效性。同时,与GCPN和MRNN在训练过程中作为自回归模型不同,GraphAF定义了一个从分子图结构到碱基分布的前馈神经网络,因此能够并行计算准确的数据似然。因此,GraphAF的训练过程是非常高效的。
我们在标准ZINC数据集上进行了广泛的实验。结果表明,graphhaf的训练效率显著,比目前最先进的GCPN模型快2倍。通过结合生成过程中的化学规则,他生成的分子是100%有效的。我们还惊奇地发现,即使在生成过程中没有使用化价检查的化学规则,GraphAF生成的有效分子的百分比仍然高达68%,显著高于现有的最先进的GCPN。这说明GraphAF确实具有较高的模型能力来学习分子结构的数据分布。我们通过强化学习进一步微调生成过程,以优化生成分子的化学性质。结果表明,在属性优化和约束属性优化任务上,GraphAF的性能明显优于之前最先进的GCPN。
相关工作
最近,人们提出了多种用于分子图生成的深度生成模型。RVAE模型使用变分自动编码器生成分子,并提出了一种新的正则化框架,以确保语义的有效性。Jin提议将分子表示为化学支架的连接树,并提出了分子产生的JT-VAE模型。对于基于虚拟空间的方法,化学性质的优化通常是通过贝叶斯优化在潜在空间中搜索来实现的。 De Cao& Kipf使用生成对抗网络进行分子生成。最先进的模型是建立在基于自回归的方法。You根据当前的子图结构动态地添加新的节点和边,将问题表述为一个连续的决策过程,并且生成策略网络由强化学习框架进行训练。最近,Popova提出了一种自回归模型MolecularRNN,基于生成的节点和边缘序列生成新的节点和边缘。自回归模型的迭代特性允许在生成过程中有效地利用化学规则进行价检查,因此这些模型生成的有效分子的比例非常高。然而,由于序列生成的性质,训练过程通常是缓慢的。我们的GraphAF方法具有自回归模型等迭代生成过程(从观测空间映射到潜空间)的优点,同时计算出前馈神经网络对应的精确似然(从观测空间映射到潜空间),它可以通过并行计算有效地实现。
最近的两项工作Graph Normalizing flow (GNF)和GraphNVP也是基于流的图生成方法。但是,我们的工作和他们的工作在精神上是完全不同的。GNF定义了一个从基本分布到预训练的Graph Autoencoders的隐藏节点表示的规范化流程。该生成方案通过两个独立的阶段完成,首先用归一化流生成节点嵌入,然后在第一阶段根据生成的节点嵌入生成图形。 相比之下,在GraphAF中,我们定义了一个从碱基分布直接到分子图结构的自回归流,可以端到端训练。GraphAF还定义了从碱基分布到分子图结构的规范化流。在我们的GraphAF中,我们将生成过程定义为一个顺序决策过程,并通过图神经网络有效地捕获子图结构,在此基础上,我们定义了一个策略函数来生成节点和边。顺序生成过程也允许结合化学规则。这样可以保证所生成分子的有效性。我们在表1中总结了现有方法。
预准备
自回归流
标准化流定义了一个参数化的可逆确定性转换,从基本分布
ε
\varepsilon
ε(潜在空间,如高斯分布)到真实世界的观测空间
Z
Z
Z(如图像和语音)。设
f
:
ε
→
Z
f : \varepsilon \rightarrow Z
f:ε→Z为一个可逆变换,其中pE()为基分布,那么我们可以通过变量变换公式计算出真实数据Z的密度函数,即
p
Z
(
Z
)
pZ(Z)
pZ(Z)。
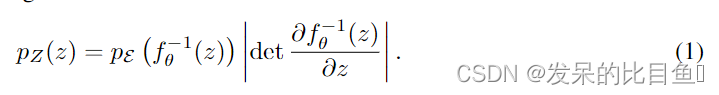
现在考虑两个关键的过程规范化流程作为一个生成模型:(1)计算数据似然:给定一个数据点
z
z
z,通过变换
f
,
ϵ
=
f
θ
−
1
(
z
)
f, \epsilon =f^{-1}_\theta(z)
f,ϵ=fθ−1(z)求逆,可以计算出精确密度
p
Z
(
z
)
pZ(z)
pZ(z)。(2)采样:
z
z
z可以通过先采样从
ϵ
∼
P
ε
(
ϵ
)
\epsilon \sim P\varepsilon(\epsilon)
ϵ∼Pε(ϵ)中的
p
Z
(
z
)
pZ(z)
pZ(z)分布中采样,然后对
z
=
f
θ
(
ϵ
)
z = f_θ(\epsilon)
z=fθ(ϵ)进行前馈变换。为了有效地执行上述运算,要求
f
θ
f_θ
fθ是可逆的,并且具有易于计算的雅可比行列式。自回归流(AF)最初在Papamakarios中提出,是满足这些条件的一种变型,它拥有一个三角形雅可比矩阵,行列式可以线性计算。形式上,给定
z
∈
R
D
z \in R^D
z∈RD(D为观测数据的维数),自回归条件概率可以参数化为高斯分布:
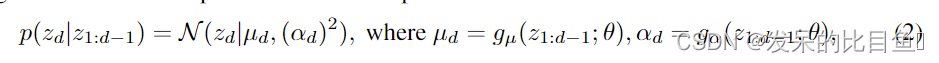
其中 g μ g_μ gμ和 g α g_α gα分别为 z 1 : d − 1 ; θ z_{1: d-1; \theta} z1:d−1;θ的无约束正标量函数,用于计算均值和偏差。在实践中,这些函数可以作为神经网络来实现。AF的仿射变换为:
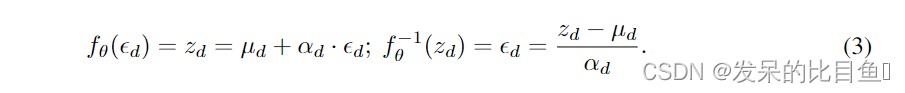
AF中的雅可比矩阵是三角形的,因为
∂
z
i
/
∂
ϵ
j
\partial{z_i}/\partial{\epsilon_j}
∂zi/∂ϵj只有对于
j
≤
i
j \leq i
j≤i是非零的。因此,可以通过
∏
d
=
1
D
α
d
\prod_{d=1}^ D \alpha_d
∏d=1Dαd有效地计算行列式。具体来说,为了进行密度估计,我们可以并行地应用所有单个标量仿射变换来计算基密度,每个基密度依赖于之前的变量
z
1
:
d
−
1
z_{1: d-1}
z1:d−1;要对
z
z
z进行采样,我们可以先对
ϵ
∈
R
D
\epsilon \in R^D
ϵ∈RD进行采样,通过仿射变换计算
z
1
z_1
z1,然后根据之前观察到的
z
1
:
d
−
1
z_{1: d-1}
z1:d−1,依次计算后面的
z
d
z_d
zd。
图表示学习
根据已有的工作,我们还将分子表示为图
G
=
(
A
,
X
)
G = (A,X)
G=(A,X),其中
A
A
A是邻接张量,
X
X
X是节点特征矩阵。假设图中有
n
n
n个节点,
d
d
d和
b
b
b分别是不同类型的节点和边的数量,则
A
∈
{
0
,
1
}
n
×
n
×
b
A \in \{0,1\}^{n \times n \times b}
A∈{0,1}n×n×b和
X
∈
{
0
,
1
}
n
×
d
X \in \{0,1\}^{n \times d}
X∈{0,1}n×d。
A
i
j
k
=
1
A_{ijk}=1
Aijk=1如果存在第
i
i
i和第
j
j
j节点之间具有
K
K
K型的键。图卷积网络(GCN)是一种用于学习图表示的神经网络体系结构。在本文中,我们使用一种关系型GCN (R-GCN)学习具有范畴边类型的图的节点表示(即原子)。设
k
k
k表示嵌入维数。我们通过聚合来自不同边缘类型的消息来计算R-GCN第
l
l
l层的节点嵌入
H
l
∈
R
n
×
k
H^l \in R^{n \times k}
Hl∈Rn×k。
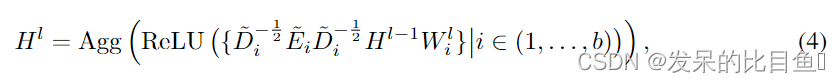
其中
E
i
=
A
[
:
,
:
,
i
]
E_i = A[:,:,i]
Ei=A[:,:,i]表示边条件邻接张量的第
i
i
i片,
E
i
~
=
E
+
I
\tilde{E_i} = E+I
Ei~=E+I,和
D
i
~
=
Σ
k
E
i
~
[
j
,
k
]
\tilde{D_i} = \Sigma_k \tilde{E_i}[j,k]
Di~=ΣkEi~[j,k]。W^{(l)}_i是第
i
i
i类边的可训练权值矩阵。
A
g
g
(
⋅
)
Agg(·)
Agg(⋅)表示一个聚合函数,如平均池或总和。将初始隐藏节点表示
H
0
H^0
H0设为原始节点特征矩阵
X
X
X。在
L
L
L消息传递层之后,我们使用最终的隐藏表示
H
L
H^L
HL作为节点表示。同时,整个图表示可以通过使用一个读出函数聚合整个节点表示来定义,如求和。
主要方法
Graphaf框架
与已有的GCPN和MolecularRNN类似,我们将分子图生成问题形式化为一个序列决策过程。设 G = ( A , X ) G = (A,X) G=(A,X)表示分子图结构。从一个空图 G 1 G_1 G1开始,每一步根据当前子图结构 G i G_i Gi生成一个新节点 X i X_i Xi,即 p ( X i ∣ G i ) p(X_i|G_i) p(Xi∣Gi)。然后,根据当前图结构,依次生成该新节点与现有节点之间的边,即 p ( A i j ∣ G i , X i , A i , 1 : j − 1 ) p(A_{ij}|G_i,X_i,A_{i,1:j-1}) p(Aij∣Gi,Xi,Ai,1:j−1)。这个过程重复,直到所有的节点和边都生成。图1(a)给出了一个示例。
GraphAF旨在定义一个从基分布(如多元高斯分布)到分子图结构
G
=
(
A
,
X
)
G = (A,X)
G=(A,X)的可逆变换。注意,我们在两个节点之间添加了另一种类型的边,这相当于两个节点之间没有边,即:
A
∈
{
0
,
1
}
n
×
n
×
(
b
+
1
)
A \in \{0,1\}^{n \times n \times (b+1)}
A∈{0,1}n×n×(b+1), 由于节点类型
X
i
X_i
Xi和边缘类型
A
i
j
A_{ij}
Aij都是离散的,不适合基于流的模型,标准的方法是使用去量化技术,通过添加实值噪声将离散数据转换为连续数据。我们采用这种方法将离散图
G
=
(
A
,
X
)
G = (A,X)
G=(A,X)预处理为连续数据
z
=
(
z
A
,
z
X
)
z = (z^A,z^X)
z=(zA,zX)
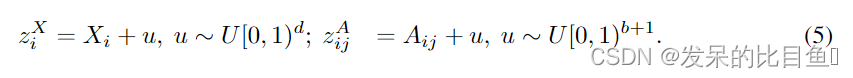
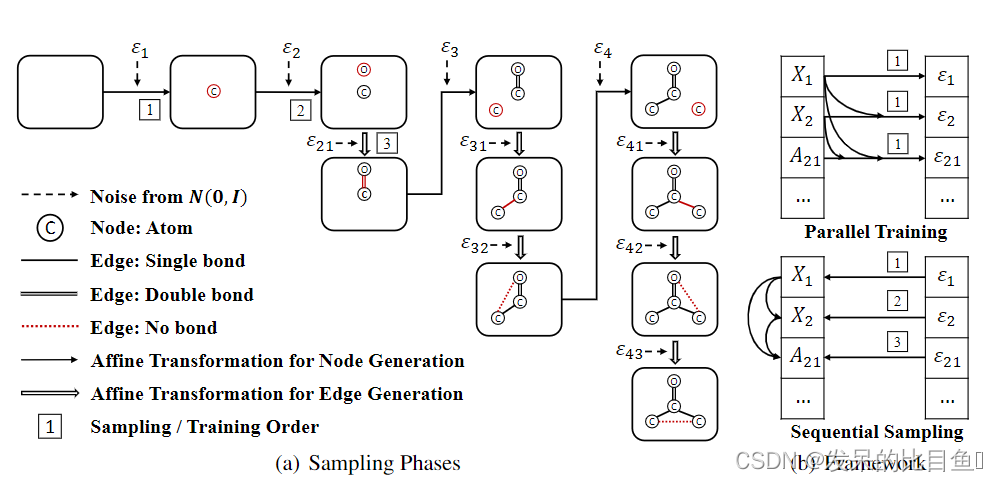
图1:GraphAF模型的概述 (a)生成过程的说明。新的节点或边用红色标记。从空图和迭代示例随机变量开始,以将它们映射到原子/键特征。编号的前三步对应于图1(b)底部图中的地图。(b)GraphAF的计算图。左边是结点和边右边是潜在变量。
我们将在附录a中进一步讨论去量化技术。我们将生成的条件分布定义为
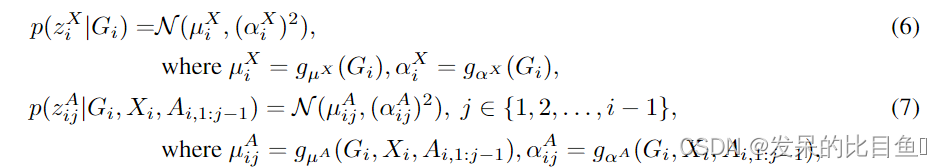
其中
g
μ
X
g_μX
gμX,
g
μ
A
g_μA
gμA和
g
α
X
g_αX
gαX,
g
α
A
g_αA
gαA是参数化的神经网络,用于定义高斯分布的均值和标准差。更具体地说,考虑到当前子图指出胃肠道结构,我们使用一个L-layer GCN(3.2节)中定义的关系来学习节点嵌入HLi Rn k,和整个嵌入的子图指出嗨Rk,基于我们定义高斯分布的均值和标准差分别生成节点和边。更具体地说,给定当前子图结构
G
i
G_i
Gi, 我们使用l层的关系型GCN(定义在3.2节)来学习节点嵌入
H
i
L
∈
R
n
×
k
H_i^L \in R^{n \times k}
HiL∈Rn×k以及整个子图的嵌入
h
i
~
∈
R
k
\tilde{h_i} \in R^k
hi~∈Rk,在此基础上定义高斯分布的均值和标准差,分别生成节点和边缘
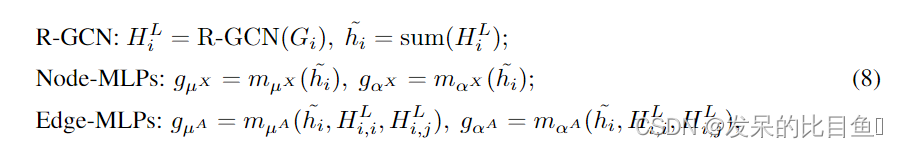
其中sum表示和池操作,而
H
i
L
∈
R
n
×
k
H_i^L \in R^{n \times k}
HiL∈Rn×k表示嵌入中第
j
j
j个节点的嵌入
H
i
L
.
m
u
x
,
m
u
x
,
m
a
x
H_i^L . m_ux, m_ux,m_ax
HiL.mux,mux,max是根据当前子图嵌入预测节点类型的多层感知器(MLP)。
为了生成一个新节点
X
i
X_i
Xi及其边缘与现有节点相连,我们只需从基高斯分布中抽样随机变量
ϵ
i
\epsilon_i
ϵi和
ϵ
i
j
\epsilon_{ij}
ϵij,并将其转换为离散特征。更准确地说
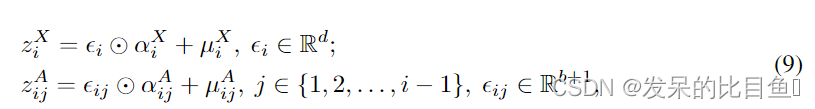
⊙ \odot ⊙是元素级乘法。在实际中,一个真实的分子图是通过取生成的连续向量的argmax来生成的,即 X i = v a r g m a x ( z x X ) d X_i = v^d_{argmax(z_x^X)} Xi=vargmax(zxX)d和 A i j = v a r g m a x ( z i j A ) b + 1 A_{ij}=v^{b+1}_{argmax(z^A_{ij})} Aij=vargmax(zijA)b+1,其中 v q p v^p_q vqp表示一个 p p p维的一维向量, q t h q^{th} qth维等于1。
令
ϵ
=
{
ϵ
1
,
ϵ
2
,
ϵ
21
,
ϵ
3
,
ϵ
31
,
ϵ
32
,
…
,
ϵ
n
,
ϵ
n
1
,
…
,
ϵ
n
,
ϵ
n
−
1
}
\epsilon=\{\epsilon_1,\epsilon_2,\epsilon_{21},\epsilon_3,\epsilon_{31},\epsilon_{32},…, \epsilon_{n}, \epsilon_{n1},…,\epsilon_{n},\epsilon_{n-1}\}
ϵ={ϵ1,ϵ2,ϵ21,ϵ3,ϵ31,ϵ32,…,ϵn,ϵn1,…,ϵn,ϵn−1},其中
n
n
n为给定分子中的原子数,GraphAF定义了一个基本高斯分布
ϵ
\epsilon
ϵ与分子结构
z
=
(
z
A
,
z
X
)
z = (z_A,z_X)
z=(zA,zX)之间的可逆映射。根据公式9,可以很容易地计算出
z
=
(
z
A
,
z
X
)
z = (z^A,z^X)
z=(zA,zX)到
ϵ
\epsilon
ϵ的逆过程为
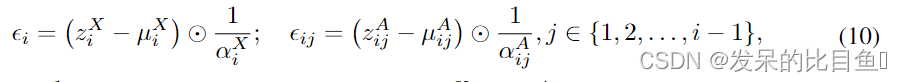
其中
1
α
i
X
\frac{1}{\alpha_i^X}
αiX1和
1
α
i
j
A
\frac{1}{\alpha^A_{ij}}
αijA1分别表示
α
i
X
\alpha_i^X
αiX和
α
i
j
A
\alpha^A_{ij}
αijA的逐元倒数
有效的并行训练
在GraphAF中,由于 f : ε → Z f:\varepsilon \rightarrow Z f:ε→Z是自回归的,所以逆过程 f − 1 : Z → ε f^{-1}:Z \rightarrow \varepsilon f−1:Z→ε的雅可比矩阵是一个三角矩阵,它的行列式可以非常有效地计算出来。给定一小批训练数据 G G G,通过式1中的变量变换公式可以有效地计算出给定顺序下每个分子的精确密度。我们的目标是最大化训练数据的可能性。
在训练过程中,我们可以利用掩蔽在输入分子图 G G G和输出潜在变量 ϵ \epsilon ϵ之间定义一个前馈神经网络来进行并行计算。掩码在推断节点 i i i的隐藏变量(即 i i i)时,去掉一些来自输入的连接,以确保R-GCN只连接子图 G i G_i Gi,即 ϵ i \epsilon_i ϵi,在推断边 A i j A_{ij} Aij的隐藏变量即 ϵ i j \epsilon_{ij} ϵij时,连接子图 G i , X i , A i , 1 : j − 1 G_i, X_i, A_{i,1:j-1} Gi,Xi,Ai,1:j−1。这与MADE和MAF中使用的方法类似。通过making技术,GraphAF满足自回归特性,同时通过并行计算所有条件, p ( G ) p(G) p(G)可以在一次前向传递中有效地计算出来。
为了进一步加速训练过程,根据广度优先搜索(BFS)顺序对训练图的节点和边进行重新排序,这种顺序是现有图生成方法中广泛采用的。由于BFS的性质,绑定只能出现在相同或连续BFS深度内的节点之间。因此,节点之间的最大依赖距离受单个BFS深度中节点数量的限制。在我们的数据集中,任何单一BFS深度都不包含超过12个节点,这意味着我们只需要对当前原子和最新生成的12个原子之间的边缘进行建模。
由于篇幅限制,我们将详细的训练算法总结在附录B中。

有效性约束抽样
在化学中,存在着许多化学规律,这些规律有助于生成有效分子。得益于顺序生成过程,GraphAF可以在每个生成步骤中利用这些规则。具体来说,我们可以明确地在抽样过程中应用价价约束来检查当前的键是否超过了允许的价价,这在以前的模型中被广泛采用。让
∣
A
i
j
∣
|A_{ij}|
∣Aij∣表示化学键
A
i
j
A_{ij}
Aij的顺序。在
A
i
j
A_{ij}
Aij的每个边生成步骤中,我们检查第
i
i
i和第
j
j
j个原子的价约束如下
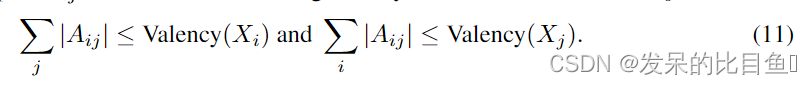
如果新加入的键打破了价约束,我们就拒绝键
A
i
j
A_{ij}
Aij,在潜在空间中抽样一个新的
ϵ
i
j
\epsilon_{ij}
ϵij,生成另一种新的键类型。当满足以下条件之一时,生成过程将终止:1)图的大小达到最大值
n
n
n, 2)新生成的原子和前一个子图之间没有生成键合。最后,把氢加到价电子未填满的原子上。
目标导向分子生成与强化学习
到目前为止,我们已经介绍了如何使用GraphAF对分子图结构的数据密度进行建模,生成有效的分子。尽管如此,对于药物的发现,我们也需要优化生成的分子的化学性质。在这一部分,我们介绍了如何微调我们的生成过程强化学习,以优化生成的分子的性质。
State and Policy Network: 状态是当前子图,初始状态是空图。该策略网络与第4.1节中定义的自回归模型相同,其中包括基于当前子图生成新原子的过程,以及生成新原子和现有原子之间的边,既 p ( X i ∣ G i ) p(X_i|G_i) p(Xi∣Gi) and p ( A i j ∣ G i , X i , A i , 1 : j − 1 ) p(A_{ij}|G_i,X_i,A_{i,1:j−1}) p(Aij∣Gi,Xi,Ai,1:j−1), 策略网络本身定义了分子图 G G G的分布 p θ p_θ pθ。如果新生成的原子和当前子图之间没有边,则生成过程终止。对于状态转移动力学,我们也加入了价位检验约束。
Reward design. 与GCPN You类似,我们也将训练策略网络的中间奖励和最终奖励结合起来。如果边的预测违反了配价检验,则引入一个小的惩罚作为中间奖励。最终的奖励包括生成的分子的靶向特性的评分,如辛醇-水分配系数(logP)或药物相似性(QED) 和化学有效性奖励,如对空间张力过大的分子和或功能基团违反锌功能基团过滤器的惩罚。最终的奖励分配到所有的中间步骤,并有一个折现因子来稳定训练。在实践中,我们采用了近端策略优化(PPO) ,一种先进的策略梯度算法,在上述定义的环境中训练GraphAF,设
G
i
j
是子图
G_{ij}是子图
Gij是子图G_i \cup X_i \cup A_{i,1:j-1}$的简写。
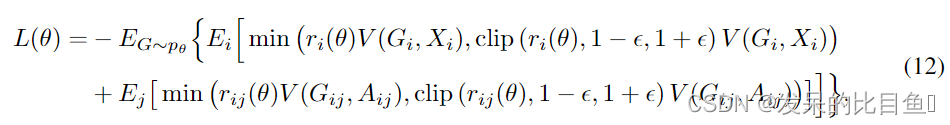
其中 r i ( θ ) = p θ ( X i ∣ G i ) p θ o l d ( x i ∣ G i ) r_i(\theta) = \frac{p_\theta(X_i|G_i)}{p\theta_{old}(x_i|G_i) } ri(θ)=pθold(xi∣Gi)pθ(Xi∣Gi)和 r i j ( θ ) = p θ ( A i j ∣ G i j ) p θ o l d ( A i j ∣ G i j ) r_{ij}(\theta) = \frac{p\theta(A_{ij}|G_{ij})}{p\theta_{old}(A_{ij}|G_{ij})} rij(θ)=pθold(Aij∣Gij)pθ(Aij∣Gij)是新旧策略输出概率的比值, V ( s t a t e , a c t i o n ) V (state,action) V(state,action)是带有移动平均基线以减少方差的估计优势函数。更具体地说,我们将生成一个节点及其与现有节点的所有边作为一个步骤,并为每个步骤维护一个移动平均基线。被剪切的代理目标可以防止策略因为一些极端的奖励而崩溃。
实验
实验设置
评估任务
在分子生成的现有工作之后,我们在三个标准任务上通过与最先进的方法进行比较进行实验。密度建模和生成评估模型学习数据分布和生成真实和多样化分子的能力。性质优化集中于生成具有优化化学性质的新分子。对于这个任务,我们微调我们的网络从密度建模任务预训练,以最大限度地提高所需的属性。约束性质优化首先由Jin等人(2018)提出,其目的是在满足相似性约束的情况下,修改给定的分子以改善所需的性质。
数据
我们使用ZINC250k分子数据集进行训练。数据集包含25万个类药物分子,最大原子数为38。它有9种原子类型和3种边缘类型。我们使用开源化学软件RDkit对分子进行预处理。除去氢后,所有分子都以kekulized的形式呈现。
Baselines
我们将GraphAF与以下分子生成的最新方法进行了比较。JT-VAE是一种基于VAE的模型,它通过先解码脚手架的树结构,然后将它们组装成分子来生成分子。JT-VAE已显示出优于其他基于VAE的模型。GCPN是一种最先进的方法,它结合了增强学习和图形表示方法,以探索庞大的化学空间。MolecularNN(MRNN)是另一种自回归模型,使用RNN以顺序产生分子。我们还将模型与最近提出的基于流动的模型GraphnVP进行了比较。除非陈述,否则基线的结果是从原始论文中获取的。
实现细节
raphAF在Pytorch中是实现。 R-GCN分为3层实现,设置嵌入维数为128。根据经验将最大图形大小设置为48。或者密度建模,我们对模型进行10个epoch的训练,批大小为32,学习速率为0.001。在属性优化方面,我们对超参数进行网格搜索,并根据化学评分性能选择最佳设置。我们使用Adam来优化我们的模型。完整的训练细节可以在附录C中找到。
表2:不同模型在密度建模和生成方面的比较。重建仅在潜在变量模型上进行评估。有效性与检查仅在具有价值约束的模型上评估。通过运行GCPN的开源代码获得†的结果。带有‡的结果取自Popova
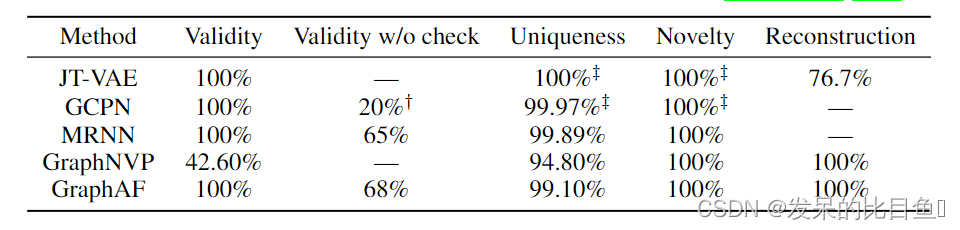
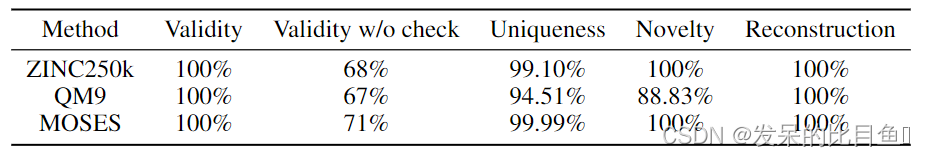
表3:在三个不同数据集上密度建模和生成的结果。
数据结果
密度建模与生成
我们通过使用广泛使用的指标来评估所提出的方法建模真实分子的能力:有效性是所有生成的图中有效分子的百分比。唯一性是所有生成的分子中唯一分子的百分比。新颖性是指生成的分子中没有出现在训练集中的百分比。重构是指潜在载体能被重构的分子的百分比。我们从10000个随机生成的分子中计算出上述指标。
表2显示,GraphAF在所有四个指标上都获得了具有竞争力的结果。GraphAF作为一种基于流的模型,与VAE方法相比具有较好的重构能力。我们的模型还实现了100%的有效性,因为我们可以在顺序生成期间利用有效性检查。而另一种基于流的方法GraphNVP,由于它的一次采样过程,其有效性仅为42.60%。一个有趣的结果是,即使在生成过程中没有有效性检查,GraphAF仍然可以达到高达68%的有效性,而之前最先进的方法GCPN只能达到20%。这表明GraphAF在建模数据密度和从大型化学数据集的无监督训练中获取领域知识方面具有很强的灵活性。我们还比较了不同方法在同一计算环境下的效率,一台拥有1 Tesla V100 GPU和32个CPU核的机器。得到表2的结果,JT-VAE和GCPN分别需要24小时和8小时左右,而GraphAF只需要4小时。
为了证明GraphAF没有过度拟合到特定的数据集ZINC250k,我们还在另外两个分子数据集QM9 和MOSES 上进行了实验。QM9包含134k分子和9个重原子,而MOSES则更大更具有挑战性,它包含1.9M分子和多达30个重原子。表3显示,即使在更复杂的数据集上,GraphAF也总能生成有效且新颖的分子。此外,虽然GraphAF最初是为分子图生成而设计的,但它实际上非常通用,只需修改节点和边生成函数edge - mlps和node - mlps。遵循图归一化流(GNF)的实验设置,我们在两个通用图数据集上对GraphAF进行了测试:COMMUNITY-SMALL,这是一个包含100个2-社区图的合成数据集;EGO-SMALL,这是一个从Citeseer数据集中提取的图集。在实际应用中,我们使用一个热点指标向量作为R-GCN的节点特征。我们借用GraphRNN的开源脚本来生成数据集并评估不同的模型。为了进行评估,我们使用You等提出的图上的一些特定指标,报告了生成图和训练图之间的最大均值差异(MMD)。从表4的结果可以看出,当应用于通用图时,GraphAF仍然可以得到与GraphRNN和GNF相当或更好的结果。我们在附录D中给出了生成的通用图的可视化。
表4:不同图生成模型在使用MMD度量的一般图上的比较。我们遵循GNF的评估方案,基线的结果也取自GNF。
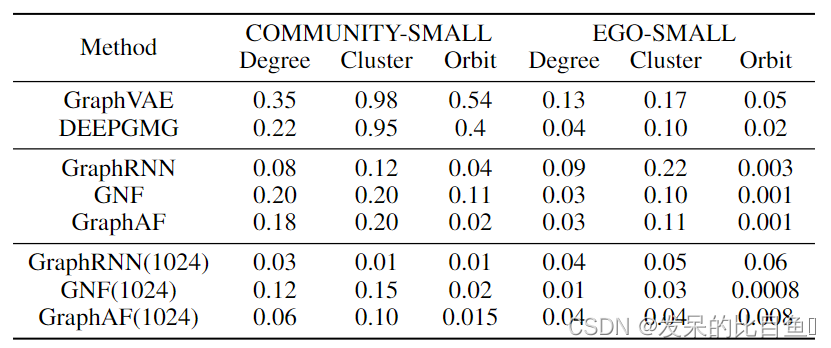
表5:生成的分子性能得分前3名的比较
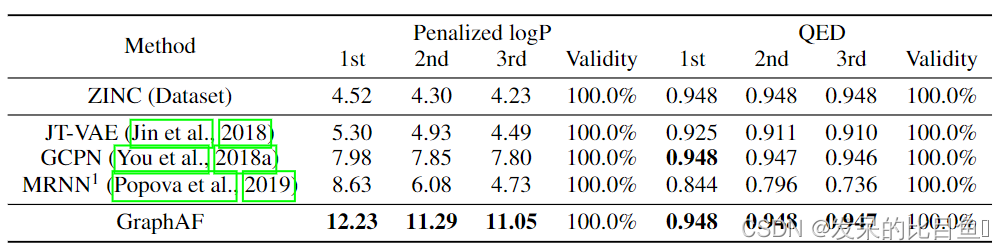
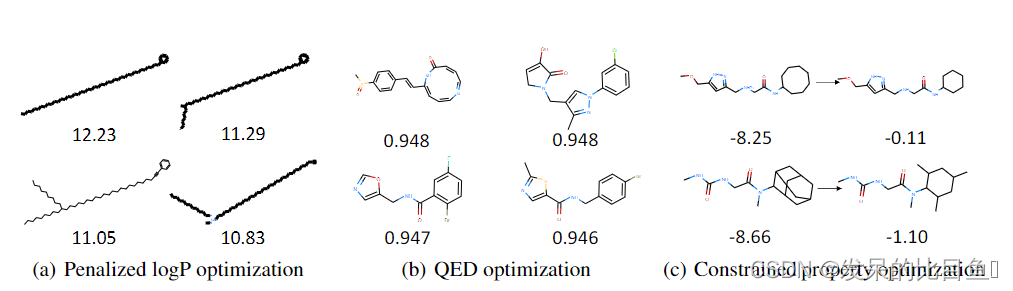
图2:性质优化和受限性质优化任务中生成的分子。(a)惩罚logP分数高的分子。(b) QED分数高的分子。(c)在受约束性质优化的惩罚性logp中,有两对分子,相似性为0.71(顶部)和0.64(底部)。
表6:约束性质优化结果比较
属性优化
在这项任务中,我们的目标是生成具有所需性质的分子。具体来说,我们选择惩罚logP和QED作为我们的目标属性。前者是由环大小和合成可及性减去的logP评分,后者是衡量分子的药物相似性。请注意,两个分数都是使用经验预测模型计算的,我们采用了在中使用的脚本,以使结果具有可比性。为了完成这项任务,我们对GraphAF网络进行300个时期的预训练,以进行似然建模,然后应用4.4节所述的RL过程,对网络进行微调,使其达到所需的化学性质。详细的奖励设计和超参数设置见附录C。在已有的工作中,我们报告每个模型的前3名得分。
如表5所示,GraphAF在惩罚logP分数上比所有基线表现得好很多,并在QED中获得了类似的结果。这一现象表明,结合RL过程,GraphAF成功捕获了所需分子的分布。需要注意的是,我们对molecular arrnn发现的前3个分子的性质进行了重新评估,结果显示它们的性质低于原文报道的结果。图2(a)和2(b)显示了我们的模型中发现的得分最高的分子。附录E中的图6展示了由GraphAF生成的更真实的分子,logP评分从5到10。
应该注意的是,正如第4.4节中定义的,我们的RL过程接近于之前的工作GCPN中使用的过程。因此,良好的性能优化性能来自于流动的灵活性。与已知存在模式崩溃问题的GCPN中使用的GAN模型相比,flow在建模复杂分布和生成不同数据方面具有灵活性(如表2和表3)。这使得GraphAF能够在RL过程中探索各种分子结构,以优化分子性能。
约束优化
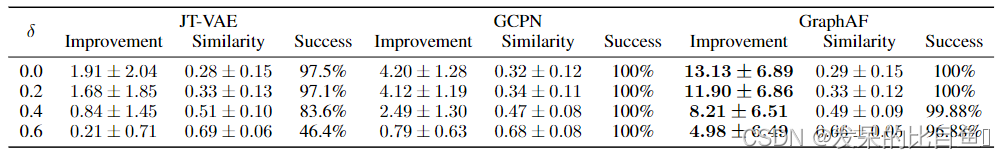
最后一项任务的目标是对给定的分子进行修饰以改善特定的性质,条件是修饰后的分子与原分子的相似度必须大于阈值δ。继Jin和You之后,我们选择优化ZINC250k中得分最低的800个分子的惩罚logP,采用Tanimoto相似度与Morgan指纹作为相似性度量。
与属性优化任务类似,我们通过密度建模对GraphAF进行预训练,然后使用RL对模型进行微调。在生成过程中,我们将初始状态设置为从800个待优化分子中随机抽样的子图。为了评价,我们在表6中报告了最高改进的平均值和标准差,以及相应的原始和改性分子之间的相似性。实验结果表明,GraphAF的性能明显优于以往的所有方法,并且几乎总是能成功地提高目标属性。 图2©可视化了两个优化示例,表明我们的模型能够在保持原始分子和改性分子之间高度相似性的同时,大幅度提高惩罚后的logP分数。
结论
我们提出了GraphAF,这是第一个基于流的自回归模型,用于生成真实和多样的分子图。由于正规化流动的灵活性,GraphAF能够模拟复杂的分子分布,并在经验实验中生成新颖的100%有效的分子。此外,GraphAF的训练是非常有效的。为了优化生成分子的性质,我们用强化学习微调生成过程。实验结果表明,在标准任务中,GraphAF的表现优于所有之前的最先进基线。未来,我们计划在更大的数据集上训练我们的graphhaf模型,并将其扩展到生成其他类型的图结构(例如,社交网络)。


















 577
577

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











