







PyAutoFEP 自由能微扰计算–基于Gromacs
简介
注意:以下教程假定读者熟悉分子动力学 (MD) 和自由能微扰 (FEP) 理论。此外,了解 GROMACS 工具、拓扑和输入文件也很有用。整个教程都使用 Linux 命令行。最后,建议用户阅读并参考 PyAutoFEP 手册,其中描述了程序的详细信息。
PyAutoFEP 使用 GROMACS 自动设置和分析小分子与大分子靶标结合的相对自由能。PyAutoFEP 自动生成扰动图、设置计算和分析。它使用灵活的 λ 方案并集成 REST2,允许对扰动区域进行部分回火,从而加快收敛速度。PyAutoFEP 具有三个模块:扰动图生成器、用于设置仿真系统和准备输入的模块以及分析模块。在本教程中,这三个模块将用于准备和分析一组扰动。

本教程中使用的模型系统是法尼醇 X 受体,一种与胆汁酸核胆汁酸受体相关的核受体,与调节胆汁分泌、胆固醇稳态和控制血糖水平 相关。由于胆汁分泌在脂质吸收中的核心作用,FXR 是与肥胖相关的代谢紊乱的药物靶标,如 2 型糖尿病。几种与不同配体共结晶的晶体结构可用于 FXR 以及结构-活性关系研究。具体来说,本教程使用由 D3R 6 (https://drugdesigndata.org/about/grand-challenge-2/fxr) 组织的社区挑战 Grand Challenge 2 中的自由能数据集 2 的一部分。


本教程中使用的螺环系列的选定分子。(使用MarvinSketch 20.3创建的图)
教程要求
-
“PyAutoFEP (see requirements in the manual or in Github: https://github.com/lmmpf/PyAutoFEP) ◦ This tutorial assumes PyAutoFEP install dir to be in path, reader may have to fix PyAutoFEP execution commands otherwise
-
GROMAC”
-
“openbabel (version 2.4.x)
-
Tutorial data (https://github.com/luancarvalhomartins/PyAutoFEP/tree/master/docs/tutorial01)”
教程数据包含输入文件。以下所有命令都假定教程数据已提取到工作目录。 PyAutoFEP 使用 GROMACS 二进制文件来准备 MD 系统和运行 MD。
大分子目标和配体输入数据
为了设置结合计算的相对自由能,PyAutoFEP 需要描述配体及其目标的输入文件。请注意,PyAutoFEP 不会参数化小分子,也不检查所提供参数的完整性、有效性或一致性。建议用户确保 (a) 每个扰动图中的所有小分子都使用一致的方法进行参数化; (b) 小分子力场和参数与选择的大分子力场兼容。
PyAutoFEP 需要配体结构(例如,mol2 或 mol 文件)和这些配体的 GROMACS 兼容拓扑(例如,top 文件)。配体结构将被读取并用于计算 MCS、排列和扰动原子,而配体拓扑将被处理,但最终由 GROMACS grompp 使用。确保配体结构中的原子顺序和原子命名与拓扑中的相匹配。目前,已经报道了与主要力场一致的小分子参数化工具 7-10。只要拓扑与 GROMACS 兼容,PyAutoFEP 就会解析由类似外部工具生成的拓扑。
配体结构和拓扑
在本教程中,LigParGen 服务器 (http://zarbi.chem.yale.edu/ligpargen; 8) 将用于获取小分子参数。为了参数化配体,首先必须生成 mol 文件。这可以使用 openbabel 来完成:
# Convert smiles in mol2 files
obabel fxr_fe2_selected_ligands.smi -Oligand.mol --gen3d -m
# Create a dir
mkdir lig_data
# Batch rename and move the mol files
for eachfile in ligand*.mol; do mv $eachfile "lig_data/$(head -n1 $eachfile).mol"; done
注意:可能最正确的方法是使用更高层次的理论来优化配体。PyAutoFEP 将使用从 mol 或 mol2 输入文件中读取的配体几何结构来准备溶剂——但不是从位姿数据中读取的复杂。建议用户验证配体结构。
接下来,LigParGen 网络服务器用于使用 lig_data 中的 .mol 文件获取配体参数。配体是单阴离子,所以电荷模型应该是“1.14*CM1A”,分子电荷是“-1”。对于每个分子,必须下载 GROMACS 兼容拓扑(GROMACS 标签下的顶部按钮)并将其作为 FXR_XX.itp 保存到 lig_data 目录,其中 FXR_XX 是配体名称。拓扑 [ moleculetype ] 指令中的配体名称将是错误的,但 PyAutoFEP 会修复它。 lig_data 目录必须包含以下文件: 最后,必须将晶体配体 FXR_10 复制到配体文件中。稍后将需要使用核心约束对齐。
蛋白质结构
# receptor_data must contain:
grep "HETATM.*9MV" receptor_data/5q17.pdb > receptor_data/9mv.pdb
PyAutoFEP 需要准备好的描述大分子的 PDB。大分子结构(例如,pdb 文件)将在配体比对过程中使用,并经过处理以添加配体。PDB 将由 GROMACS 处理,因此它必须包含氢,并且残基和原子名称必须符合 GROMACS 力场规范。在本教程中,将使用 GROMACS 兼容的 OPLS-AA/M 力场(力场数据可在此处获取(链接:http://zarbi.chem.yale.edu/oplsaam.html),oplsaam.ff 目录应解压到工作目录。
PDB2PQR 服务器 (http://server.poissonboltzmann.org/pdb2pqr; 11) 是一种广泛使用的准备 PDB 文件的工具,它方便地集成了 PropKa 程序以预测残留物的滴定状态。在本教程中,PDB2PQR 网络服务器将用于修复 5Q17 (ref) pdb 文件(模型缺失原子,删除备用位置和共结晶介质种类)并将氢添加到测定 pH (7.4)。输出中必须使用 AMBER 命名方案。输出 pdb 必须在 receptor_data 目录中保存为 5q17_processed.pdb。
注意:如果使用命令行 pdb2pqr,可以使用以下命令准备 PDB 文件:“pdb2pqr30 --ffout=AMBER --titration-state-method=propka --with-ph=7.4 --protonate-all --drop-water 5Q17 5q17_processed.pdb” (pdf)
然而,会有一些命名有问题的残基。为了本教程的目的,重命名残基和原子的 bash 脚本与数据一起可用。
# Replace residue names
./rename_res.sh receptor_data/5q17_processed.pdb
rename_res.sh 是一个简单的脚本,使用 sed 就地编辑 receptor_data/5q17_processed.pdb 并在 receptor_data/5q17_processed.pdb.bak 中创建备份。
注意:本教程中包含 rename_res.sh 以简化其管道,它不是 PyAutoFEP 的一部分。它不适用于任意 PDB 文件和力场。用户有责任确保输入的 PDB 与使用的力场一致。
扰动图生成
获得一组相对结合能的第一步是生成扰动图。扰动图是表示一组分子中的扰动的连通图,因此每个顶点代表一个分子,每条边代表一个扰动。
**注意:**自动地图生成是可选的,自定义地图可以直接提供给第二个模块。
**注意:**地图生成器将构建连接图,但这不是 PyAutoFEP 工作的要求;可以使用断开连接的地图,但并非所有分析都可用。
扰动图生成器可以准备三种类型的扰动图。在本教程中,将使用更简单的星图。星图是一种扰动图,其中所有配体都连接到一个中心配体,以便产生尽可能少的扰动。在扰动图的设置过程中,PyAutoFEP 将计算中心配体和所有其他配体之间的最大公共子结构 (MCS),并且此信息将被保存以供以后重复使用。 MCS 和其他数据将保存到进度文件中,这是一个可以由 PyAutoFEP 读取的 Python3 pickle 文件。
要使用 FXR_12 作为中心分子生成星图,可以使用以下命令。它将读取 lig_data 中的所有 .mol 文件,计算 MCS 并将扰动图保存到 progress.pkl。地图的图形描述将保存到 best_graph.svg。
# Generate a star map generate_perturbation_map.py --map_type=star --map_bias=FXR_12 --input lig_data/*.mol

准备输入
PyAutoFEP 的主要部分 prepare_dual_topology.py 创建对偶拓扑、设置 MD 系统并准备 GROMACS 输入。将生成目录结构运行脚本,以便使用单个脚本运行所有内容。运行脚本可以直接运行 GROMACS gmx mdrun 或将作业提交给调度程序,例如 Slurm 或 PBS。
为了准备复杂的legs,prepare_dual_topology.py 需要输入关于 FXR 的配体姿势。通常,此类姿势可以通过无约束对接、约束核心对接或实验结构获得。然而,在本教程中,将使用一种更简单的方法。 PyAutoFEP 可以通过将输入配体叠加到参考小分子结构来准备输入姿势,只要系列和参考分子共享一个共同的核心。
注意:当通过叠加生成姿势时,PyAutoFEP 不会考虑大分子,并且将使用简单的构象采样策略,因此叠加仅在扰动较小且刚性时才可行。对于更多样化的系列,核心约束对接策略可能是更好的选择。
另一个相关点是为最小化、平衡和运行扰动而生成的脚本类型。默认选项将生成一个通用的串行 bash 脚本,该脚本将串行运行每个扰动腿。为了简化向作业调度程序的提交,PyAutoFEP 可以制作将提交作业的脚本,而不是直接运行它们。
注意:使用串行 bash 脚本可能不是一个好主意,即使对于本教程中的一些扰动也是如此。 PyAutoFEP 手册描述了如何设置 prepare_dual_topology.py 来准备运行脚本以将作业提交给 SLURM、Torque 或 PBS。
最后,在教程的这一部分,将使用配置文件而不是使用命令行参数。这简化了输入,尤其是当必须设置许多选项时。 PyAutoFEP 可以读取 ini 文件作为配置,命令行中可用的所有参数也可作为配置选项使用(有关更多信息,请参见手册)。
这些是配置文件的内容:
# 这是 prepare_dual_topology.py 的配置部分(这是此文件中唯一的部分,但是 [ section ] 是强制性的
[prepare_dual_topology]
# 从此文件夹中读取配体和拓扑结构
input_ligands = lig_data
# 从这个文件中读取大分子结构
structure = receptor_data/5q17_processed.pdb
# 这是力场目录,它将被复制到每个扰动目录并用于准备MD系统
extradirs = oplsaam.ff
# 控制核心约束叠加的选项
# 首先选择使用它而不是读取所有配体姿势
pose_loader = superimpose
# 使用这个pose作为叠加的参考
poses_reference_pose_superimpose = receptor_data/9mv.pdb
# 输出名称。这将是一个自解压的 bash 文件
perturbations_dir = tutorial
# 在运行节点中设置 GROMACS 可执行文件的路径,如果需要取消注释和修改。
# gmx_bin_run = /usr/local/bin/gromacs
# 控制输出的选项,更多信息参见手册。根据需要取消注释
# FEP 腿将被提交给 slurm 调度程序
# output_scripttype = slurm
# 在慢跑开始时运行这些命令(对于加载模块、导入库很有用)
# output_runbefore = module load python3; module load cuda
# 微调作业资源
# output_resources = all_cpus:24; all_gpus:2; all_time: 24
# 在收集步骤中使用 python 文件而不是二进制文件
# output_collecttype = python
然后可以使用以下命令运行 prepare_dual_topology.py 脚本:
# 准备系统
prepare_dual_topology.py --config_file=step2.ini --output_hidden_temp_dir=False
`prepare_dual_topology.py` 将运行片刻并创建一个 tutorial.bin 文件。
运行 FEP
将 tutorial.bin 复制到将运行计算的集群或机器后,必须将其提取。
# The default is a self-extracting script, but see manual of other options
bash ./tutorial.bin
将创建一个具有完整目录结构的教程目录,以运行所有 FEP legs并收集数据。
在这个目录中有一个方便的脚本,tutorial.bin,用于运行所有内容(如果使用 output_scripttype = bash,默认),或将所有legs作为作业提交(如果 output_scripttype = slurm)。
# Run or submit all
cd tutorial
ls # Contents may vary
FXR_12-FXR_74 FXR_12-FXR_84 FXR_12-FXR_88 pack.sh runall.sh FXR_12-FXR_76 FXR_12-FXR_85 collect_results_from_xvg progress.pkl
bash runall.sh
注意:将数据收集到 FEP 所需的 MD 在集群节点中可能需要几个小时。建议用户确保有足够的计算资源可用。
执行 runall.sh 将使用串行 bash 脚本直接运行 MD。无论是否使用作业调度器,运行过程都是相似的。每个系统的每个 λ 窗口将被最小化和平衡。然后,使用 GROMACS multirun,将运行所有 λ 窗口的 MD。 MD 完成后,GROMACS 重新运行将用于使用所有 λ 窗口的哈密尔顿重新评估轨迹。最后,将使用 collect_results_from_xvg 收集 MD 数据。
注意:有关运行过程的详细说明,请参阅 PyAutoFEP 手册。
脚本运行完成或所有作业完成后,教程目录中将出现一个新文件 tutorial.tgz。此文件包含 PyAutoFEP 分析所需的所有数据,应从集群或远程计算机复制回来,以防使用其中任何一个。
分析
PyAutoFEP 简化了 FEP 数据的分析,计算了 ΔΔG 并为每个扰动生成了几个有用的图。 PyAutoFEP 完成的一些分析使用 alchemical-analysis.py。强烈建议用户参考此参考资料以获得有关自由能计算分析的精彩讨论。
注意:PyAutoFEP 通过 alchemlyb 使用 pymbar 来获得 ΔΔG。确保您使用的是 pymbar >= 3.0.4,因为较新的版本有一个可能导致错误的错误(https://github.com/choderalab/pymbar/issues/419)
运行 analyze_results.py 所需的唯一文件是 tutorial.tgz 输出。可以使用以下命令运行分析:
# Run all analysis, use kcal/mol as unit, and calculate ΔΔG relative to the center
# molecule FXR_12 (--center_molecule=FXR_12 is redundant in the case and could be omitted)
analyze_results.py --input tutorial.tgz --units kcal --output_uncompress_directory tutorial --center_molecule=FXR_12
# analyse_results.py will print some data, including the formatted pairwise ΔΔG
这将创建一个教程目录,其中包含一些文件和每个扰动的目录。所有分子相对于中心分子的 ΔΔG 将写入 ddg_to_center.csv。从这个文件中,可以生成一个关联实验和计算的图。可以在 gc2_data/PyAutoFEP 教程 plots.ipynb Jupyter notebook 中找到如何执行此操作的示例,它使用来自 gc2_data/experimental_affinity.csv 的实验数据。

来自 gc2_data/PyAutoFEP 教程 plots.ipynb 的相关图。深灰色区域为 1 kcal/mol 误差范围。浅灰色是 2 kcal/mol 误差范围。对角虚线是理论上的 Y=X 相关性。
在每个扰动目录中,水和蛋白质目录包含每条腿的数据。这可用于评估预测的质量。例如,ddg_vs_time.svg 文件将包含在轨迹评估到特定时间时估计的 ΔΔG 图。该图可用于评估 FEP 的收敛性,因为收敛良好的估计将在接近完整模拟时具有稳定的估计 ΔΔG。此外,对于更长的模拟时间,使用反向轨迹估计的 ΔΔG 应该与常规(正向)轨迹相似。可以对模拟进行扩展以达到收敛。
ΔΔG 在连续时间使用截断轨迹估计。请注意,正向和反向轨迹的预测值彼此非常相似,并且在 1.5 ns 后随时间稳定。
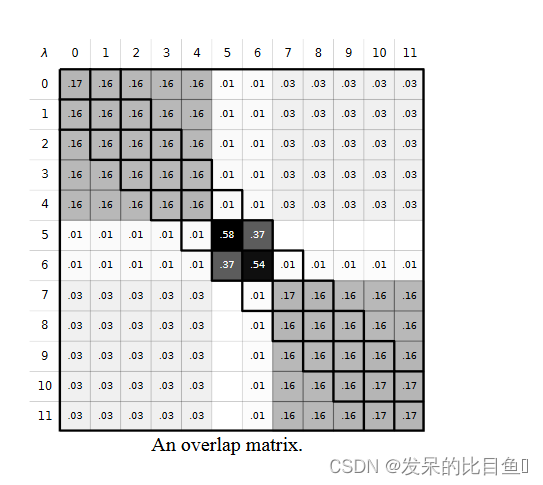
overlap_matrix.svg 将包含状态之间重叠的估计值。良好的重叠要求第一个上下对角线至少为 0.03。不同的 λ 方案可以帮助改善重叠(请参阅手册以了解有关在 PyAutoFEP 中选择 λ 方案的更多信息)。

此外,一个名为 analysis 的目录将与包含每个 lambda MD(lambdaX,其中 X 是整数)的数据的目录一起出现。在这个分析目录中,对于复杂的扰动,将有包含配体和蛋白质的 RMSD、RMSF、SASA 和距离数据的 xvg 文件。这些可用于检查轨迹的特征并评估配体稳定性。请注意,所有中间状态(即除终点外的所有状态)都是非物理状态,因此建议用户在解释此类状态的数据时要谨慎。
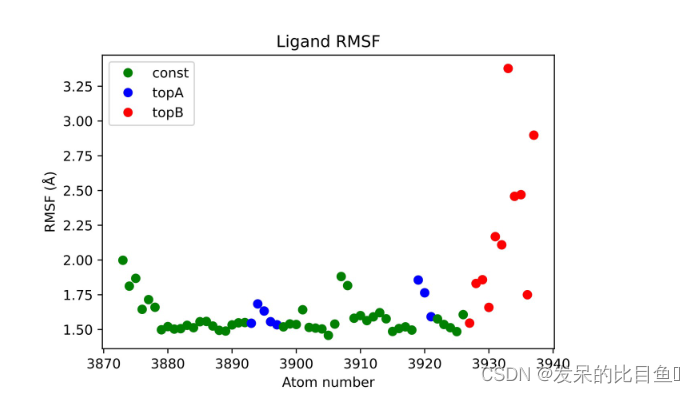
状态 λ = 0 的配体 RMSF。请注意,拓扑结构 B 原子(红色)是非相互作用的粒子,其 RMSF 没有物理意义。 (使用 Python3、numpy 和 matplotlib 准备的绘图)。















 4695
4695











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











