









基于Gromacs配体修饰自由能FPE计算(手动版)
本教程来自于https://github.com/huichenggong/Learning-Computation-with-Chenggong/tree/main/CC_news_008_ddG_uniFEP
我们将要使用的系统来自这篇论文
配体和受体pdb文件
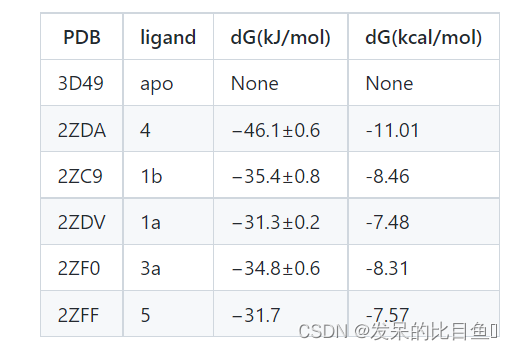
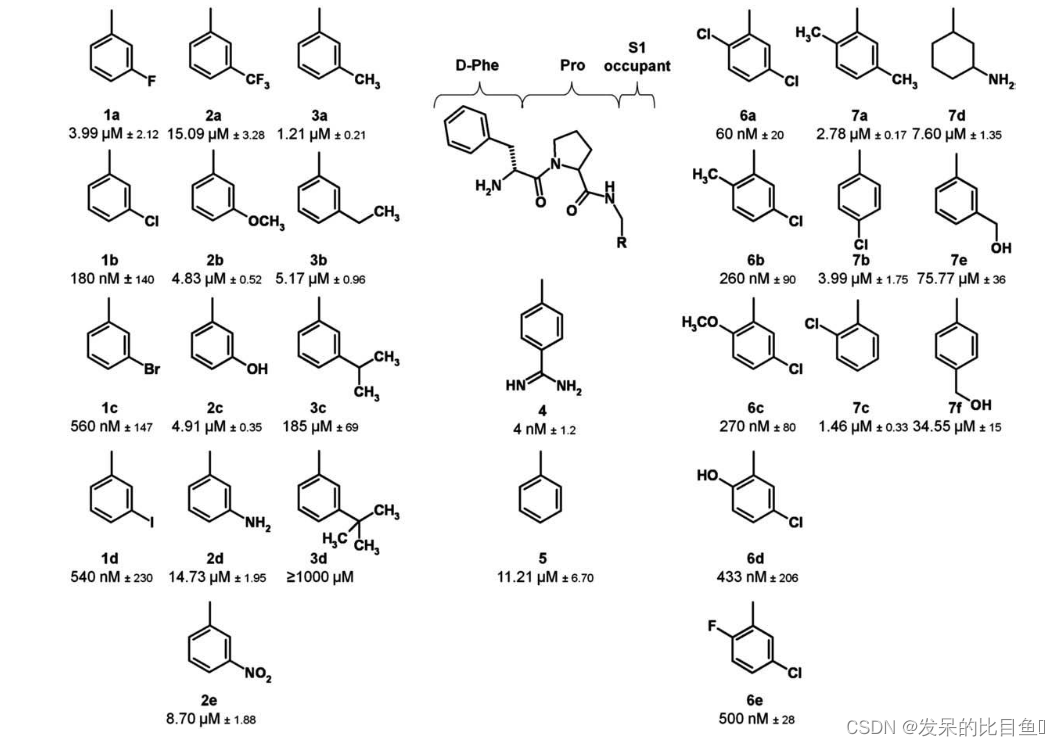
A. 介绍
在本教程中,我们将使用非平衡自由能计算装置来估计与凝血酶结合的两个配体之间的相对自由能差。双重自由能差可以根据下面描述的热力学循环来表示
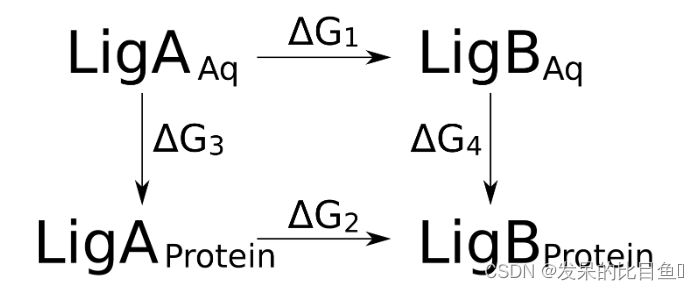
在这种情况下,我们感兴趣的是得到配体B和A的结合自由能之差,即
Δ
G
4
−
Δ
G
3
ΔG_4 - ΔG_3
ΔG4−ΔG3。
Δ
Δ
G
=
Δ
G
4
−
Δ
G
3
=
Δ
G
2
−
Δ
G
1
ΔΔG = ΔG_4 - ΔG_3 = ΔG_2 - ΔG_1
ΔΔG=ΔG4−ΔG3=ΔG2−ΔG1
这样,我们将用更可行的炼金术配体变形从状态A到B。这种炼金术修饰我们将以非平衡的方式进行。我们将使用PMX设置过渡的所需结构和拓扑。对于配体结构,处理PMX依赖于RDKIT软件包。
B. 混合结构/拓扑结构
首先,我们将为后续的模拟生成混合结构和拓扑。在本教程中,我们将考虑本出版物中描述的一组配体中的两种配体:配体1a和配体5,如图1所示。对于这两种分子,已经按照Amber力场(GAFF)的标准程序制备了拓扑结构:lig 1a和lig 5。在文件夹中,您将找到一个.pdb格式的配体结构文件和两个拓扑文件:MOL.itp and ffMOL.itp. 包含原子、键、角和二面体的信息,而原子类型(范德华参数)则被分离到ffMOL.itp文件中。为了进一步使用,可以方便地将两个原子类型的ffMOL.itp文件合并为一个(one_ff_file.py脚本将执行合并)
python one_ff_file.py -ffitp lig_1a/ffMOL.itp lig_5/ffMOL.itp -ffitp_out ffMOL.itp
为了生成混合结构和拓扑结构,首先,我们将映射lig_1a和lig_5的原子,以确定哪些原子可以在分子之间发生变形。这个过程将使用pmx包。原子映射由脚本atoms_to_morph.py执行到
python atoms_to_morph.py -i1 lig_1a/lig_1a.pdb -i2 lig_5/lig_5.pdb -o pairs.dat -alignment -H2H
以上的程序将根据每个原子间距离进行对齐后的映射。此外,氢原子也会变成氢原子(而不是把所有的氢原子都变成假人)。可以随意使用映射选项,例如,标记-mcs将基于配体的最大公共子结构执行基于拓扑的映射,然而,对于当前配体对,结果保持不变。该脚本的输出是映射的分数(out_score.dat):接近于零的数字表示配对中的配体彼此相似,适合进行炼金术修饰。另一个输出是原子的实际映射:pairs.dat文件。接下来,我们将使用创建的原子映射来生成配体对的混合结构和拓扑(make_hybrid.py脚本)
python make_hybrid.py -l1 lig_1a/lig_1a.pdb -l2 lig_5/lig_5.pdb -itp1 lig_1a/MOL.itp -itp2 lig_5/MOL.itp -pairs pairs.dat -oa merged.pdb -oitp MOL.itp -ffitp ffmerged.itp -scDUMd 0.0
新创建的merged.pdb文件包含混合结构(请注意原子列表末尾的虚拟原子)。merged.pdb中的坐标基于脚本提供的第一个配体,即lig_1a。mergedB.pdb具有与merged.pdb相同的原子,但其中的坐标基于lig_5。提供的MOL.itp文件包含了为两个物理状态明确定义的所有参数的混合拓扑结构。混合结构所需的任何虚拟原子在ffmerged.itp拓扑文件中定义。让我们将此原子类型文件与先前生成的ffMOL.itp合并:
python one_ff_file.py -ffitp ffMOL.itp ffmerged.itp -ffitp_out ffMOL.itp
对于进一步的模拟,我们还将需要蛋白质的拓扑:凝血酶。我们可以通过运行标准gromacs工具PDB2GMX(我们将使用Amber99SB-Star-Uldn-Mut Force Field)来获得它:
gmx pdb2gmx -f thrombin.pdb -o thrombin_pdb2gmx.pdb -water none -ff amber99sb-star-ildn-mut
此命令将生成具有添加的氢原子的凝血酶结构。此外,还将创建三个拓扑文件,分别命名为topol_Protein_chain_*.itp,用于结构文件中的每个蛋白链。将所有所需的.itp文件组装到.top文件中是方便的。根据上面图片中的热力学循环,我们需要设置两个不同的仿真分支:1)配体在水中;2)配体结合蛋白质。对于这两种情况,我们已经准备好了.top文件:top_water.top和top_protein.top。生成所有所需的拓扑后,我们可以在下一步按照标准Gromacs仿真设置程序进行操作。
C. 平衡模拟
下面的模拟设置步骤可以很容易地添加到脚本中,以方便地实现自动化过程。然而,在本教程中,我们将明确地介绍这些步骤,以提供设置的完整详细图片。
需要四组平衡模拟:
- 1)水中配体状态a,
- 2)水中配体状态b,
- 3)与蛋白质结合的配体状态a,
- 4)与蛋白质结合的配体状态b。
因此,可以方便地分别创建4个目录
mkdir stateA_water
mkdir stateB_water
mkdir stateA_protein
mkdir stateB_protein
溶剂化模拟盒只需要为水和蛋白质中的“stateA”准备。对于“stateB”,我们将重新使用相同的模拟设置,并通过.mdp参数简单地更改状态。首先,将杂化配体结构放在一个盒子里
gmx editconf -f merged.pdb -o stateA_water/box.pdb -d 2 -bt dodecahedron
对于与蛋白质结合的配体,我们需要找到配体在凝血酶活性位点的最佳姿势。有几种方法可以做到这一点:对接,MD模拟,对准实验确定的结构。在目前的情况下,初始配体坐标是基于晶体学凝血酶结构和结合抑制剂(pdb ID: 2ZDA)构建的。这样,混合结构已经处于合适的姿态。将配体和蛋白质加在一起,生成盒子
awk '/ATOM/ {print $0}' thrombin_pdb2gmx.pdb merged.pdb > stateA_protein/thrombin_ligand.pdb
gmx editconf -f stateA_protein/thrombin_ligand.pdb -o stateA_protein/box.pdb -d 2 -bt dodecahedron
溶剂化系统(我们明确定义了添加的水的数量,以便与将在进一步步骤中使用的预先计算的轨迹兼容)
gmx solvate -cs spc216.gro -cp stateA_water/box.pdb -o stateA_water/water.pdb -p top_water.top -maxsol 3729
gmx solvate -cs spc216.gro -cp stateA_protein/box.pdb -o stateA_protein/water.pdb -p top_protein.top -maxsol 23720
要添加离子,首先下载.mdp文件(它们也将在后面的设置步骤中使用)。注意,我们将使用由jung and Cheatham重新参数化的非标准离子:NA, CL。在这种情况下,要添加的离子的数字明确地提供了与进一步使用的预先计算的轨迹的兼容性。添加的离子中和模拟盒,达到150毫米盐浓度。
gmx grompp -f mdp/emA.mdp -c stateA_water/water.pdb -o stateA_water/water.tpr -p top_water.top -maxwarn 1
gmx grompp -f mdp/emA.mdp -c stateA_protein/water.pdb -o stateA_protein/water.tpr -p top_protein.top -maxwarn 2
echo 4 | gmx genion -s stateA_water/water.tpr -nn 10 -np 10 -o stateA_water/ions.pdb -nname CL -pname NA -p top_water.top
echo 15 | gmx genion -s stateA_protein/water.tpr -nn 64 -np 65 -o stateA_protein/ions.pdb -nname CL -pname NA -p top_protein.top
现在我们准备文件开始能量最小化
gmx grompp -f mdp/emA.mdp -c stateA_water/ions.pdb -o stateA_water/em.tpr -p top_water.top -maxwarn 1
gmx grompp -f mdp/emB.mdp -c stateA_water/ions.pdb -o stateB_water/em.tpr -p top_water.top -maxwarn 1
gmx grompp -f mdp/emA.mdp -c stateA_protein/ions.pdb -o stateA_protein/em.tpr -p top_protein.top -maxwarn 1
gmx grompp -f mdp/emB.mdp -c stateA_protein/ions.pdb -o stateB_protein/em.tpr -p top_protein.top -maxwarn 1
要执行能量最小化,请进入相应的目录并运行命令(假设计算机至少有4个核心,否则请调整-ntomp选项)
gmx mdrun -s em.tpr -c emout.gro -v -ntomp 4
最后,准备平衡运行
gmx grompp -f mdp/eqA.mdp -c stateA_water/emout.gro -o stateA_water/eq.tpr -p top_water.top -maxwarn 1
gmx grompp -f mdp/eqB.mdp -c stateB_water/emout.gro -o stateB_water/eq.tpr -p top_water.top -maxwarn 1
gmx grompp -f mdp/eqA.mdp -c stateA_protein/emout.gro -o stateA_protein/eq.tpr -p top_protein.top -maxwarn 1
gmx grompp -f mdp/eqB.mdp -c stateB_protein/emout.gro -o stateB_protein/eq.tpr -p top_protein.top -maxwarn 1
每个平衡模拟都设定为生成10 ns的轨迹。这样的模拟可能需要一些时间,因此我们已经提前执行了模拟,您可以下载生成的轨迹。要进一步学习本教程,请将轨迹放置在工作文件夹的相应目录中。
平衡自己运行则如下操作:
gmx mdrun -deffnm eq -v
D. 非平衡跃迁和分析
非平衡过渡将从平衡模拟中提取的结构开始。让我们从预先计算的轨迹中提取快照
echo 0 | gmx trjconv -s stateA_water/eq.tpr -f trajectories/stateA_water/traj_comp.xtc -o stateA_water/frame.gro -b 1 -pbc mol -ur compact -sep
echo 0 | gmx trjconv -s stateB_water/eq.tpr -f trajectories/stateB_water/traj_comp.xtc -o stateB_water/frame.gro -b 1 -pbc mol -ur compact -sep
echo 0 | gmx trjconv -s stateA_protein/eq.tpr -f trajectories/stateA_protein/traj_comp.xtc -o stateA_protein/frame.gro -b 1 -pbc mol -ur compact -sep
echo 0 | gmx trjconv -s stateB_protein/eq.tpr -f trajectories/stateB_protein/traj_comp.xtc -o stateB_protein/frame.gro -b 1 -pbc mol -ur compact -sep
然后,为每个起始结构生成一个.tpr文件,用于从stateA到stateB的炼金法转换,反之亦然。由于要遍历100个起始结构,因此可以方便地将以下命令放在一个脚本中,其中循环将在帧上运行(我们将以第一帧为例)
gmx grompp -f mdp/tiA.mdp -p top_water.top -c stateA_water/frame0.gro -o stateA_water/tpr0.tpr -maxwarn 1
gmx grompp -f mdp/tiB.mdp -p top_water.top -c stateB_water/frame0.gro -o stateB_water/tpr0.tpr -maxwarn 1
gmx grompp -f mdp/tiA.mdp -p top_protein.top -c stateA_protein/frame0.gro -o stateA_protein/tpr0.tpr -maxwarn 1
gmx grompp -f mdp/tiB.mdp -p top_protein.top -c stateB_protein/frame0.gro -o stateB_protein/tpr0.tpr -maxwarn 1
接下来,对每个生成的.tpr文件执行gmx mdrun -deffnm tpr0 -v命令。最有可能的情况是,您希望提交这些作业以在集群上并行运行。或者,您可以在一台快速机器上依次运行所有这些作业(可能使用GPU)。无论哪种方式,模拟都可能需要一些时间,因此,我们已经执行了所有的运行并收集了所需的输出。为了计算配体在水中的A态和B态之间的自由能差,创建一个分析文件夹(例如分析水),导航到它并运行脚本
analyze_dhdl.py -fA ../dhdlA_water/dhdl*xvg -fB ../dhdlB_water/dhdl*xvg -t 298
为了分析与蛋白质结合的配体的过渡,首先在主工作文件夹中创建一个各自的分析目录并导航到它。然后运行分析脚本
analyze_dhdl.py -fA ../dhdlA_protein/dhdl*xvg -fB ../dhdlB_protein/dhdl*xvg -t 298
所获得的results.dat文件提供了一些具有各自误差的自由能估计。其中一个常用的估计量是基于观测得到的工作分布的最大似然,被称为BAR(Benett Acceptance Ratio)。蛋白质中配体的BAR ΔG值与水中配体的BAR ΔG值的差值正是我们感兴趣的ΔΔG值。计算值为0.26 kJ/mol,而出版物中的ITC值为0.4 kJ/mol。为了更好地感觉与计算出的自由能相关的不确定性研究不同的估计器(CGI,BAR,JARZYNSKI),错误估计,收敛量度,视觉检查工作分布及其重叠。
参考
http://pmx.mpibpc.mpg.de/sardinia2018_tutorial2/index.html
















 1万+
1万+











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











