






欢迎关注”生信修炼手册”!
在NAD+代谢相关的文献中,使用了两批illumina beadchip的芯片数据进行分析,本文以其中一篇数据为例,详细展示该平台的数据处理流程。
GSE112676包含741个样本的全血基因表达谱数据,链接如下
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE112676
该数据的处理流程在以下文献中有详细描述
https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-019-1909-0

可以分为以下几步
1. 下载GenomeStudio导出的数据
GenomeStudio是处理illumina原始芯片的软件,在数据库中提供了该批数据的导出结果
该文件的内容如下
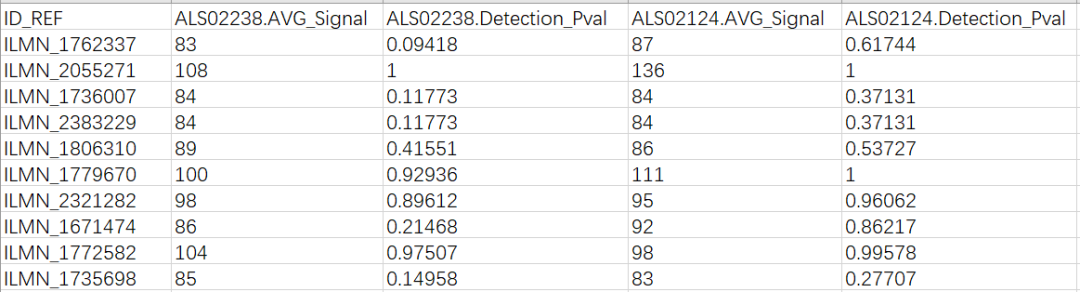
每一行为一个探针,每个样本用两列表示,第一列是AVG_Signal, 表示探针的荧光信号强度,第二列为Detection_Pval, 表示检测信号的p值。
2. 进行pvalue 的校正
计算荧光信号强度与检测p值的相关性,代码如下
> x <- read.table("GSE112676_HT12_V3_preQC_nonnormalized.txt", header = T, sep = "\t", row.names = 1)
> sample_cnt <- ncol(x) / 2
> # 计算pvalue 和 intensity 之间的相关性
> spearman_cor <- unlist(lapply(1:sample_cnt, function(t){
+ res <- cor.test(x[[t * 2 - 1]], x[[t * 2]], method="spearman")
+ res$estimate
+ }))
There were 50 or more warnings (use warnings() to see the first 50)
>
> # 统计相关系数的分布
> length(spearman_cor[spearman_cor > 0.9])
[1] 221
> length(spearman_cor[spearman_cor < -0.9])
[1] 520可以看到,正如文章中所说,520个样本的相关性小于-0.9, 221个样本的相关性大于0.9, 整体样本分为明显的两类,一类正相关,一列负相关。为了使整体保持一致,将占比较少的正相关样本的p值,改为1-P, 代码如下
> # 校正p值
> for(t in which(spearman_cor > 0.9)) {
+ x[[t * 2]] <- 1 - x[[t * 2]]
+ }
> # 校正后重新查看相关系数的分布
> spearman_cor <- unlist(lapply(1:sample_cnt, function(t){
+ res <- cor.test(x[[t * 2 - 1]], x[[t * 2]], method="spearman")
+ res$estimate
+ }))
There were 50 or more warnings (use warnings() to see the first 50)
>
>
> length(spearman_cor[spearman_cor > 0.9])
[1] 0
> length(spearman_cor[spearman_cor < -0.9])
[1] 741可以看到,校正之后,所有的样本都为负相关。
3. 背景校正和归一化
文献中描述的方法如下

使用limma包进行处理,背景校正选择normexp方法,归一化选择quantile方法,代码如下
> # 读取 illumina beadchip, 读取校正后的数据
> RG <- read.ilmn("GSE112676_HT12_V3_preQC_nonnormalized.adjust.pvalue.txt", ctrlfiles = NULL)
Reading file GSE112676_HT12_V3_preQC_nonnormalized.adjust.pvalue.txt ... ...
> # 背景校正 normal–exponential convolution model
> RG <- backgroundCorrect(RG, method="normexp")
Array 1 corrected
Array 2 corrected
Array 3 corrected
....
Array 739 corrected
Array 740 corrected
Array 741 corrected
> # quantile 归一化
> RG <- normalizeBetweenArrays(RG, method="quantile")
> dim(RG)
[1] 48803 741预处理之后,得到了741个样本共48803个探针水平的表达量。
4. 提取基因水平的表达量
由于一个基因对应多个探针,在该文献中,只使用表达量最高的探针作为该基因的表达量。以上就是一个完整的illumina芯片的数据处理流程。
·end·
—如果喜欢,快分享给你的朋友们吧—
原创不易,欢迎收藏,点赞,转发!生信知识浩瀚如海,在生信学习的道路上,让我们一起并肩作战!
本公众号深耕耘生信领域多年,具有丰富的数据分析经验,致力于提供真正有价值的数据分析服务,擅长个性化分析,欢迎有需要的老师和同学前来咨询。
更多精彩
写在最后
转发本文至朋友圈,后台私信截图即可加入生信交流群,和小伙伴一起学习交流。
扫描下方二维码,关注我们,解锁更多精彩内容!

一个只分享干货的
生信公众号















 3938
3938











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









