






 本文详细介绍了基因芯片数据分析的过程,涉及Affymetrix、Illumina和Agilent三种主要芯片的数据处理。主要内容包括芯片数据的来源、格式以及使用R包如affy、oligo、limma、beadarray、lumi等进行数据读取、质量控制、背景矫正和标准化的方法。同时,提到了各类原始数据文件,如CEL、idat、txt等,以及对应的处理工具和数据库资源。
本文详细介绍了基因芯片数据分析的过程,涉及Affymetrix、Illumina和Agilent三种主要芯片的数据处理。主要内容包括芯片数据的来源、格式以及使用R包如affy、oligo、limma、beadarray、lumi等进行数据读取、质量控制、背景矫正和标准化的方法。同时,提到了各类原始数据文件,如CEL、idat、txt等,以及对应的处理工具和数据库资源。

芯片数据分析笔记【03】 | GEO数据库使用教程及在线数据分析工具
芯片数据分析笔记【04】 | ArrayExpress 数据库介绍
芯片原始数据文件包括:① 芯片图像扫描得到的记录光信号强度的Intensity文件 ② 包含芯片类型、探针排布等芯片具体设计信息的Design文件 ③ 包含探针注释信息、探针序列等信息的Annotation文件 ④ 包含样本分组、实验处理等信息的Targets文件。不同芯片厂商的文件格式不同,下面是三大厂商芯片数据相应文件的格式信息:

原始芯片数据的来源:① GEO、ArrayExpres等多个存储芯片实验数据的数据库 ② 芯片公司的官方网站。
处理这些数据会用到下面这些R包:
affy/oligo;beadarray/lumi;limma ;AgiMicroRna ;GEOquery ;readr/readxl。
(1). Affymetrix芯片
Affymetrix芯片原始数据最常用的格式是CEL格式,从读取原始数据到转化为基因表达量矩阵表格,可以使用affy ,oligo等R包进行数据的质量控制,背景矫正,数据标准化,其中标准化方法常用的为rma算法和mas5算法。
1). affy包
R包affy用于读取Affymetrix芯片CEL文件,也可用于读取一些较早期的affy芯片数据,如3'IVT 芯片,常见的U133系列(GPL570、GPL57K GPL96) ,不适合用于读取较新的affy芯片类型,如Exon ST、 GeneSK SNP芯片。affy读取CEL文件函数是read.affybatch/ReadAffy 读取后的对象类别:AffyBatch
2). oligo包
oligo读取Affymetrix芯片CEL文件,也可以读取affymetrix的几乎所有芯片类型的CEL文件,此外,oligo还可读取Roche NimbleGen芯片的.xys原始数据文件 ,oligo读取CEL文件函数是read.celfiles,读取后的对象类别:
芯片数据 | 对象 |
Expresssion arrays | ExpressionFeatureSet |
Gene ST arrays | GeneFeatureSet |
Exon ST arrays | ExonFeatureSet |
SNP arrays | SnpFeatureSet |
Tiling arrays | TilingFeatureSet |
(2). Illumina芯片
Illumina芯片数据分析的4个起点:① Pixel-level,每个像素点对应一个数据, tif/tiff 格式;② Bead-level,每个bead对应一个数据, 类似于affymetrix的单个probe数据, 具体数据包括txt/idat/Iocs /sdf/ xml等多种格式;③ Summary-level,每个bead type对应 —个数据,类似于affy的probe set数据, txt/CSV 格式;④ 公共数据库存储的表达矩阵数据,如 GEO/ArrayExpress,部分数据集已经过预处理,属于summary-level。
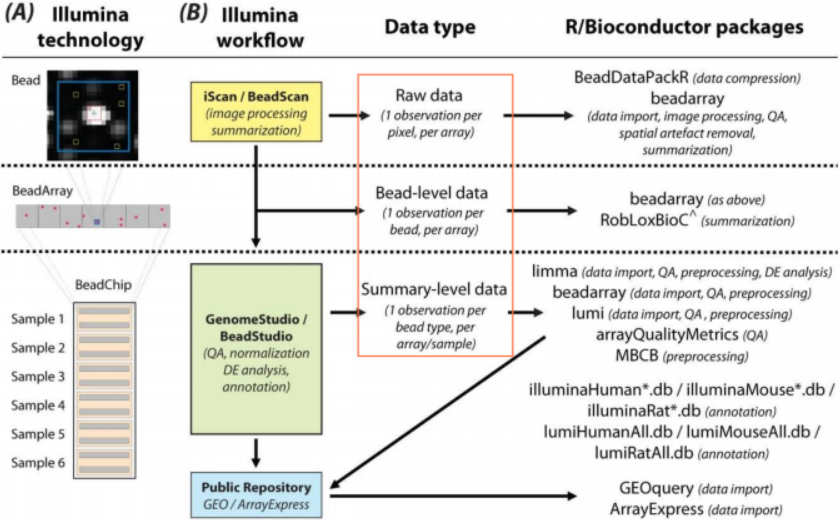
使用iScan/BeadScan扫描图像及处理图像信息时,因软件的具体版 本和设置参数不同,产生多种不同格式的原始数据文件。① .txtfiles,包含所有beads坐标数据和已减去背景值的光信号强度值 ② .tiffiles,扫描后得到的图像文件 ③ .Iocs files,包含所有beads的坐标信息 ④ .idatfiles,专有的二进制文件用于储存光信号强度数据,可供 BeadStudio/GenomeStudio读取进行后续分析 ⑤ .xml files,包含仪器的扫描参数设置和提取光强度值数据的具体方法信息 ⑥ .bgx files,包含探针注释信息 ⑦ Metrics.txt,包含扫描芯片的质量参数信息 ⑧ .sdf files,包含样本/芯片的具体构架、布局信息 ⑨ IBS (Illumina Bead Summary) files,.csv格式,summary-level 数据。
1). limma包
read.ilmn函数读取summary-level数据 , read.idat读取idat数据。
2). beadarray包
readlllumina函数最常用,用于读取bead-level数据 ;readTIFF函数读取pixel-level图像数据 ;readldatFiles函数读取idat文件;readLocsFile函数读取.Iocs文件;readBeadSummaryData函数读取summary-level数据。
3). lumi包
lumiR函数读取summary-level数据 lumiR.batch函数批量读取summary-level数据
4). illuminaio包
readIDAT函数读取idat数据 ;readBGX函数读取.bgx文件。
(3). Agilent芯片
1). limma
limma包中的read.maimages函数不限于Agilent芯片数据,可用于读取多个平台芯片文件,但不适用于affymetrix及illumina芯片数据 ,可读取单色及双色芯片数据;可读取多种图像分析程序产生的txt数据,如:Agilent Feature Extraction, Arrayvision, BlueFuse, GenePix, ImaGene, QuantArray (Version 3 or later), Stanford Microarray Database (SMD),SPOT。
2). AgiMicroRna
readMicroRnaAFE 函数适用于Agilent的miRNA芯片数据。
|
|
|
|
|
|
|
|
| |

























 4963
4963

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









