







甲基化分析应知应会的另一个R包:minfi,ChMAP包的很多的函数都有minfi包的影子。
minfi使用起来也很简单,就是5个对象走一遍流程即可,下面还是用之前的GSE194282数据集进行演示!
读取数据
suppressMessages(library(minfi))
和ChAMP包差不多,也是指定文件夹,文件夹内有IDAT文件和样本信息csv文件。
baseDir <- "./gse149282/GSE149282_RAW/"
首先是读取csv文件,这个文件需要自己制作,可以参考这篇文章:xxxxxxxxxx
targets <- read.metharray.sheet(baseDir,
pattern = "sample_type.csv"
)
## [read.metharray.sheet] Found the following CSV files:
## [1] "./gse149282/GSE149282_RAW/sample_type.csv"
targets
## Sample_Name Sample_Type Array Slide
## 1 GSM4495491 normal R01C01 GSM4495491_200811050117
## 2 GSM4495492 cancer R02C01 GSM4495492_200811050117
## 3 GSM4495493 normal R03C01 GSM4495493_200811050117
## 4 GSM4495494 cancer R04C01 GSM4495494_200811050117
## 5 GSM4495495 normal R05C01 GSM4495495_200811050117
## 6 GSM4495496 cancer R06C01 GSM4495496_200811050117
## 7 GSM4495497 normal R07C01 GSM4495497_200811050117
## 8 GSM4495498 cancer R08C01 GSM4495498_200811050117
## 9 GSM4495499 normal R01C01 GSM4495499_200811050116
## 10 GSM4495500 cancer R02C01 GSM4495500_200811050116
## 11 GSM4495501 normal R03C01 GSM4495501_200811050116
## 12 GSM4495502 cancer R04C01 GSM4495502_200811050116
## 13 GSM4495503 normal R05C01 GSM4495503_200811050116
## 14 GSM4495504 cancer R06C01 GSM4495504_200811050116
## 15 GSM4495505 normal R07C01 GSM4495505_200811050116
## 16 GSM4495506 cancer R08C01 GSM4495506_200811050116
## 17 GSM4495507 cancer R01C01 GSM4495507_202193490061
## 18 GSM4495508 normal R02C01 GSM4495508_202193490061
## 19 GSM4495509 cancer R03C01 GSM4495509_202193490061
## 20 GSM4495510 normal R04C01 GSM4495510_202193490061
## 21 GSM4495511 cancer R05C01 GSM4495511_202193490061
## 22 GSM4495512 normal R06C01 GSM4495512_202193490061
## 23 GSM4495513 cancer R07C01 GSM4495513_202193490061
## 24 GSM4495514 normal R08C01 GSM4495514_202193490061
## Basename
## 1 ./gse149282/GSE149282_RAW/GSM4495491_200811050117_R01C01
## 2 ./gse149282/GSE149282_RAW/GSM4495492_200811050117_R02C01
## 3 ./gse149282/GSE149282_RAW/GSM4495493_200811050117_R03C01
## 4 ./gse149282/GSE149282_RAW/GSM4495494_200811050117_R04C01
## 5 ./gse149282/GSE149282_RAW/GSM4495495_200811050117_R05C01
## 6 ./gse149282/GSE149282_RAW/GSM4495496_200811050117_R06C01
## 7 ./gse149282/GSE149282_RAW/GSM4495497_200811050117_R07C01
## 8 ./gse149282/GSE149282_RAW/GSM4495498_200811050117_R08C01
## 9 ./gse149282/GSE149282_RAW/GSM4495499_200811050116_R01C01
## 10 ./gse149282/GSE149282_RAW/GSM4495500_200811050116_R02C01
## 11 ./gse149282/GSE149282_RAW/GSM4495501_200811050116_R03C01
## 12 ./gse149282/GSE149282_RAW/GSM4495502_200811050116_R04C01
## 13 ./gse149282/GSE149282_RAW/GSM4495503_200811050116_R05C01
## 14 ./gse149282/GSE149282_RAW/GSM4495504_200811050116_R06C01
## 15 ./gse149282/GSE149282_RAW/GSM4495505_200811050116_R07C01
## 16 ./gse149282/GSE149282_RAW/GSM4495506_200811050116_R08C01
## 17 ./gse149282/GSE149282_RAW/GSM4495507_202193490061_R01C01
## 18 ./gse149282/GSE149282_RAW/GSM4495508_202193490061_R02C01
## 19 ./gse149282/GSE149282_RAW/GSM4495509_202193490061_R03C01
## 20 ./gse149282/GSE149282_RAW/GSM4495510_202193490061_R04C01
## 21 ./gse149282/GSE149282_RAW/GSM4495511_202193490061_R05C01
## 22 ./gse149282/GSE149282_RAW/GSM4495512_202193490061_R06C01
## 23 ./gse149282/GSE149282_RAW/GSM4495513_202193490061_R07C01
## 24 ./gse149282/GSE149282_RAW/GSM4495514_202193490061_R08C01
然后就是根据csv文件读取IDAT文件:
RGset <- read.metharray.exp(targets = targets,
force = T
)
RGset
## class: RGChannelSet
## dim: 1051815 24
## metadata(0):
## assays(2): Green Red
## rownames(1051815): 1600101 1600111 ... 99810990 99810992
## rowData names(0):
## colnames(24): GSM4495491_200811050117_R01C01
## GSM4495492_200811050117_R02C01 ... GSM4495513_202193490061_R07C01
## GSM4495514_202193490061_R08C01
## colData names(6): Sample_Name Sample_Type ... Basename filenames
## Annotation
## array: IlluminaHumanMethylationEPIC
## annotation: ilm10b4.hg19
现在列名很长,把名字改一下:
sampleNames(RGset) <- targets$Sample_Name
RGset
## class: RGChannelSet
## dim: 1051815 24
## metadata(0):
## assays(2): Green Red
## rownames(1051815): 1600101 1600111 ... 99810990 99810992
## rowData names(0):
## colnames(24): GSM4495491 GSM4495492 ... GSM4495513 GSM4495514
## colData names(6): Sample_Name Sample_Type ... Basename filenames
## Annotation
## array: IlluminaHumanMethylationEPIC
## annotation: ilm10b4.hg19
这个RGChannelSet是minfi中一个很重要的对象,类似于表达谱芯片数据中的ExpressionSet对象,接下来的一系列操作都是从这个对象开始的。β值矩阵和样本信息都在这个对象里。
beta.m <- getBeta(RGset)
## Loading required package: IlluminaHumanMethylationEPICmanifest
beta.m[1:4,1:4]
## GSM4495491 GSM4495492 GSM4495493 GSM4495494
## cg18478105 0.02875489 0.06142969 0.03667535 0.05015362
## cg09835024 0.02900064 0.03217275 0.05820881 0.09295855
## cg14361672 0.70473730 0.57060400 0.72168755 0.45141877
## cg01763666 0.82418887 0.84666806 0.66462600 0.81427334
pd <- pData(RGset) # 获取样本信息
pd[1:4,1:4]
## DataFrame with 4 rows and 4 columns
## Sample_Name Sample_Type Array Slide
## <character> <character> <character> <character>
## GSM4495491 GSM4495491 normal R01C01 GSM4495491_200811050..
## GSM4495492 GSM4495492 cancer R02C01 GSM4495492_200811050..
## GSM4495493 GSM4495493 normal R03C01 GSM4495493_200811050..
## GSM4495494 GSM4495494 cancer R04C01 GSM4495494_200811050..
甲基化矩阵的两种注释包:
- manifest:主要包含matrix design,
- annotation:甲基化位点的位置,SNP信息等。
annotation(RGset)
## array annotation
## "IlluminaHumanMethylationEPIC" "ilm10b4.hg19"
数据探索
主要是看样本质量如何。
可以画density plot:
densityPlot(dat = RGset, sampGroups = pd$Sample_Type)
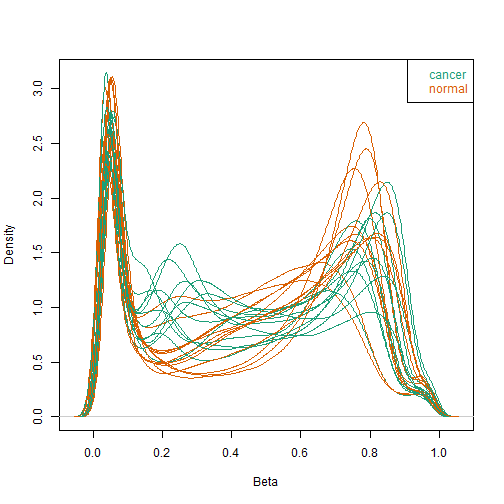
和下面这个函数一样的结果:
# 会在当前目录下生成一个pdf文件
qcReport(rgSet = RGset,
sampNames = pd$Sample_Name,
sampGroups = pd$Sample_Type,
pdf = "qcReport.pdf",
maxSamplesPerPage = 24
)
mds plot也是很简单,这个图其实就是主成分分析的图,横纵坐标分别是第一主成分、第二主成分:
mdsPlot(dat = RGset,
sampGroups = pd$Sample_Type,
sampNames = pd$Sample_Name
)
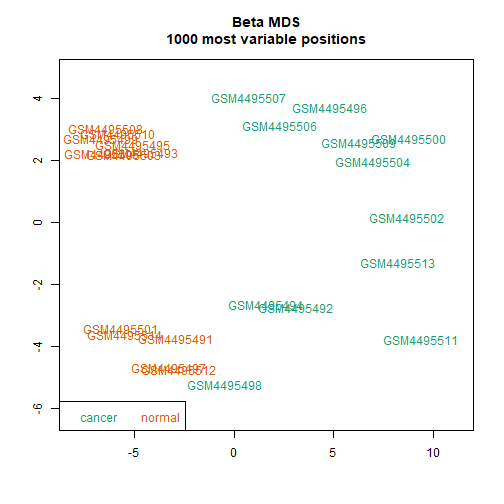
这两个图和ChAMP出的图一模一样!因为ChAMP调用了这个包的方法。
densityBeanPlot(dat = RGset,
sampGroups = pd$Sample_Type
)
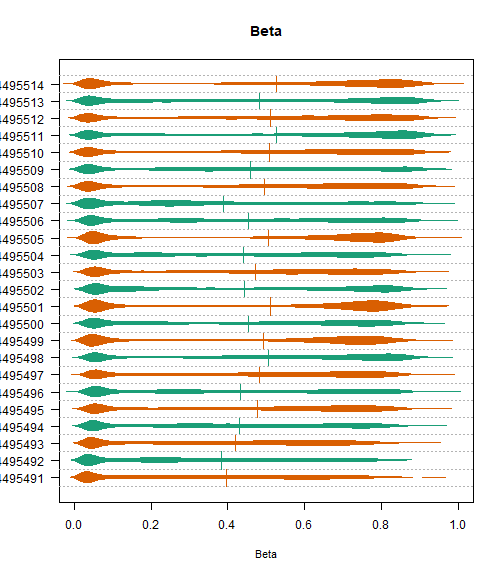
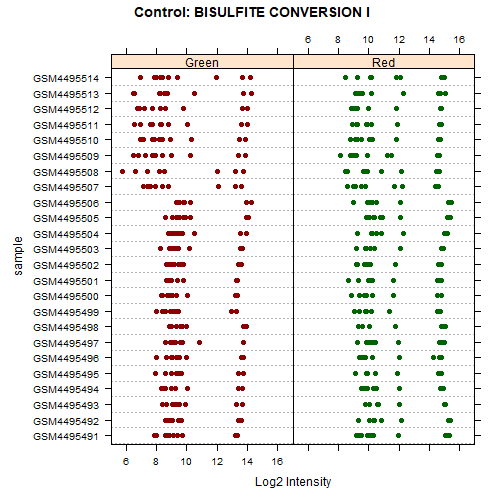
controlStripPlot(RGset)
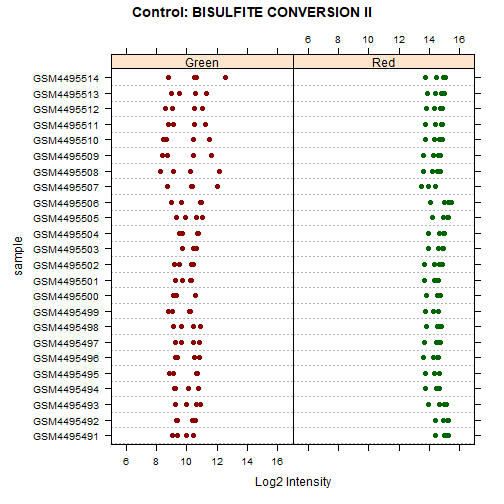
质控
首先是根据P值过滤低质量的探针和样本,一般P值大于0.05的样本要去除。
# 计算P值
detP <- detectionP(rgSet = RGset)
dim(detP)
## [1] 866091 24
detP[1:4,1:4]
## GSM4495491 GSM4495492 GSM4495493 GSM4495494
## cg18478105 0.000000e+00 0 0.000000e+00 0
## cg09835024 0.000000e+00 0 0.000000e+00 0
## cg14361672 0.000000e+00 0 0.000000e+00 0
## cg01763666 2.517235e-299 0 1.969474e-278 0
可以画图看下每个样本中的平均P值,base r就很简单了:
barplot(colMeans(detP),
col = palette.colors(24,"Alphabet"),
ylim = c(0,0.0005),
las = 2
)
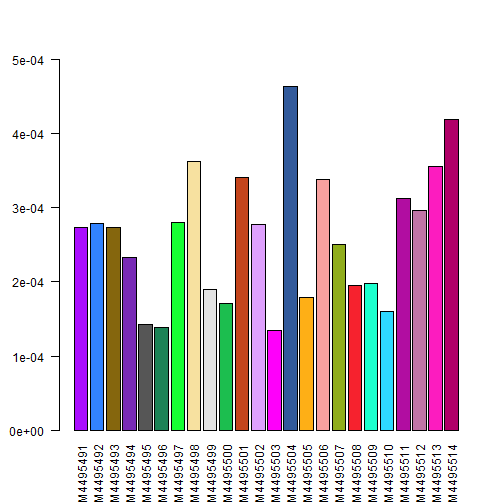
我们这个数据所有样本质量都挺好的,所以没有要去除的样本:
# 去除低质量的样本
keep <- colMeans(detP) <0.05
RGset <- RGset[, keep]
dim(RGset) # 还是24个样本
## [1] 1051815 24
记得把样本信息和P值矩阵也更新一下:
targets <- targets[keep,] # 还是24个样本
detP <- detP[,keep]
标准化
然后是对数据进行标准化。
提供了6种预处理的方法,实际使用中选择1种即可,也可以都试一下看看区别。
- preprocessRaw : No processing.
- preprocessIllumina : Illumina preprocessing, as performed by Genome Studio (reverse engineered by us).
- preprocessSWAN : SWAN normalization, described in (Maksimovic, Gordon, and Oshlack 2012).
- preprocessQuantile : Quantile normalization (adapted to DNA methylation arrays), described in (Touleimat and Tost 2012, @minfi)
- preprocessNoob : Noob preprocessing, described in (Triche et al. 2013).
- preprocessFunnorm : Functional normalization as described in (Fortin et al. 2014).
我们这个甲基化芯片是Illumina EPIC的,不同方法都试一下。
mset.raw <- preprocessRaw(rgSet = RGset)
mSet.illu <- preprocessIllumina(rgSet = RGset)
mset.swan <- preprocessSWAN(rgSet = RGset)
mset.quan <- preprocessQuantile(RGset)
## [preprocessQuantile] Mapping to genome.
## Loading required package: IlluminaHumanMethylationEPICanno.ilm10b4.hg19
## [preprocessQuantile] Fixing outliers.
## [preprocessQuantile] Quantile normalizing.
mset.noob <- preprocessNoob(rgSet = RGset)
mset.fun <- preprocessFunnorm(rgSet = RGset)
## [preprocessFunnorm] Background and dye bias correction with noob
## [preprocessFunnorm] Mapping to genome
## [preprocessFunnorm] Quantile extraction
## [preprocessFunnorm] Normalization
上面几种标准化方法有的需要基因组探针的位置,有的不需要,所以经过标准化之后变成了不同的对象:
sapply(list(mset.raw,mSet.illu,mset.swan,mset.quan,mset.noob,mset.fun),class)
## [1] "MethylSet" "MethylSet" "MethylSet" "GenomicRatioSet"
## [5] "MethylSet" "GenomicRatioSet"
前面带Genomic的是已经比对到基因组上的,这几个对象可以通过函数转化:
mSet.illu.g <- mSet.illu |>
ratioConvert() |> # 这两个函数顺序可以互换
mapToGenome()
mSet.illu.g
## class: GenomicRatioSet
## dim: 865859 24
## metadata(0):
## assays(3): Beta M CN
## rownames(865859): cg14817997 cg26928153 ... cg07587934 cg16855331
## rowData names(0):
## colnames(24): GSM4495491 GSM4495492 ... GSM4495513 GSM4495514
## colData names(6): Sample_Name Sample_Type ... Basename filenames
## Annotation
## array: IlluminaHumanMethylationEPIC
## annotation: ilm10b4.hg19
## Preprocessing
## Method: Illumina, bg.correct = TRUE, normalize = controls, reference = 1
## minfi version: 1.42.0
## Manifest version: 0.3.0
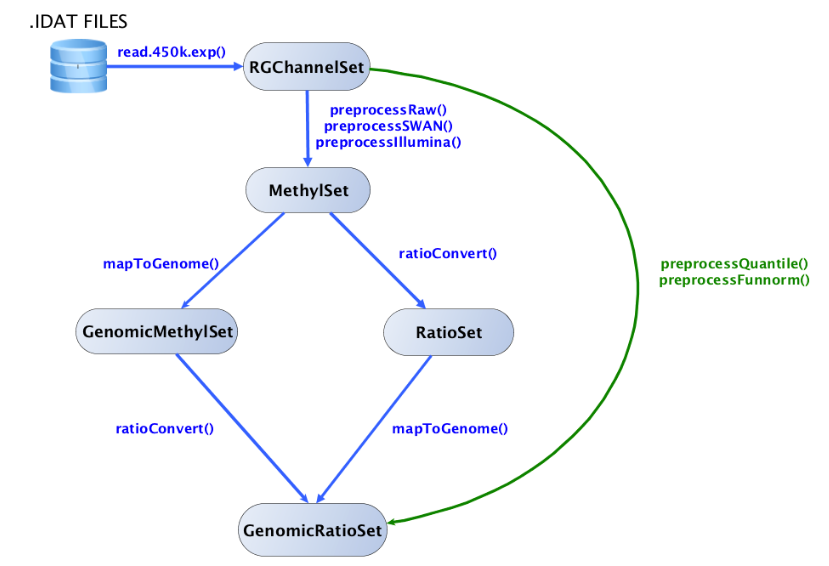
minfi中最重要的5种对象,都是从RGChannelSet开始的,它们之间的关系可以用下面这张图展示:
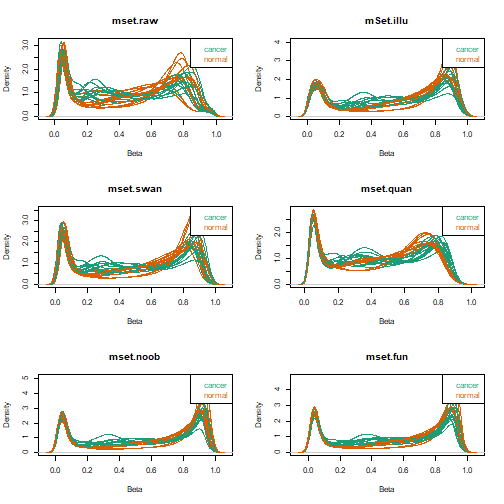
可以画图看一下使用不同的方法之后的density plot:
par(mfrow = c(3,2))
densityPlot(getBeta(mset.raw),sampGroups = pd$Sample_Type,main = "mset.raw")
densityPlot(getBeta(mSet.illu),sampGroups = pd$Sample_Type,main = "mSet.illu")
densityPlot(getBeta(mset.swan),sampGroups = pd$Sample_Type,main = "mset.swan")
densityPlot(getBeta(mset.quan),sampGroups = pd$Sample_Type,main = "mset.quan")
densityPlot(getBeta(mset.noob),sampGroups = pd$Sample_Type,main = "mset.noob")
densityPlot(getBeta(mset.fun),sampGroups = pd$Sample_Type,main = "mset.fun")
过滤
我们选择preprocessIllumina方法得到的结果继续进行接下来的分析。
首先是去除低质量的探针,P值大于0.01的探针需要去除。
# 探针顺序变一致
detP <- detP[match(featureNames(mSet.illu.g),rownames(detP)),]
# 只保留P值小于0.01的探针
keep <- rowSums(detP < 0.01) == ncol(mSet.illu.g)
table(keep)
## keep
## FALSE TRUE
## 7176 858683
去掉了7176个探针!
mSet.illu.g <- mSet.illu.g[keep,]
mSet.illu.g
## class: GenomicRatioSet
## dim: 858683 24
## metadata(0):
## assays(3): Beta M CN
## rownames(858683): cg26928153 cg16269199 ... cg07587934 cg16855331
## rowData names(0):
## colnames(24): GSM4495491 GSM4495492 ... GSM4495513 GSM4495514
## colData names(6): Sample_Name Sample_Type ... Basename filenames
## Annotation
## array: IlluminaHumanMethylationEPIC
## annotation: ilm10b4.hg19
## Preprocessing
## Method: Illumina, bg.correct = TRUE, normalize = controls, reference = 1
## minfi version: 1.42.0
## Manifest version: 0.3.0
然后是过滤掉X/Y染色体上的探针。
# 获取探针注释信息
annEPIC <- getAnnotation(IlluminaHumanMethylationEPICanno.ilm10b4.hg19)
annEPIC[1:4,1:4]
## DataFrame with 4 rows and 4 columns
## chr pos strand Name
## <character> <integer> <character> <character>
## cg18478105 chr20 61847650 - cg18478105
## cg09835024 chrX 24072640 - cg09835024
## cg14361672 chr9 131463936 + cg14361672
## cg01763666 chr17 80159506 + cg01763666
drop <- (featureNames(mSet.illu.g) %in% annEPIC$Name[annEPIC$chr %in%
c("chrX","chrY")])
table(drop)
## drop
## FALSE TRUE
## 839801 18882
mSet.illu.g <- mSet.illu.g[!drop,] # 去掉18882个探针
mSet.illu.g
## class: GenomicRatioSet
## dim: 839801 24
## metadata(0):
## assays(3): Beta M CN
## rownames(839801): cg26928153 cg16269199 ... cg07660283 cg09226288
## rowData names(0):
## colnames(24): GSM4495491 GSM4495492 ... GSM4495513 GSM4495514
## colData names(6): Sample_Name Sample_Type ... Basename filenames
## Annotation
## array: IlluminaHumanMethylationEPIC
## annotation: ilm10b4.hg19
## Preprocessing
## Method: Illumina, bg.correct = TRUE, normalize = controls, reference = 1
## minfi version: 1.42.0
## Manifest version: 0.3.0
去除对CpG有影响的SNP探针:
mSetSqFlt <- dropLociWithSnps(mSet.illu.g)
mSetSqFlt # 839801→811152
## class: GenomicRatioSet
## dim: 811152 24
## metadata(0):
## assays(3): Beta M CN
## rownames(811152): cg26928153 cg16269199 ... cg07660283 cg09226288
## rowData names(0):
## colnames(24): GSM4495491 GSM4495492 ... GSM4495513 GSM4495514
## colData names(6): Sample_Name Sample_Type ... Basename filenames
## Annotation
## array: IlluminaHumanMethylationEPIC
## annotation: ilm10b4.hg19
## Preprocessing
## Method: Illumina, bg.correct = TRUE, normalize = controls, reference = 1
## minfi version: 1.42.0
## Manifest version: 0.3.0
还需要去除映射到多个位置上面的探针,cross-reactive probes,使用的是Chen et al发现的信息。
# 需要安装methylationArrayAnalysis包
dataDirectory <- system.file("extdata", package = "methylationArrayAnalysis")
xReactiveProbes <- read.csv(file=paste(dataDirectory, "48639-non-specific-probes-Illumina450k.csv", sep="/"), stringsAsFactors=FALSE)
keep <- !(featureNames(mSetSqFlt) %in% xReactiveProbes$TargetID)
table(keep)
## keep
## FALSE TRUE
## 24675 786477
又过滤掉24675个探针!
mSetSqFlt <- mSetSqFlt[keep,]
mSetSqFlt
## class: GenomicRatioSet
## dim: 786477 24
## metadata(0):
## assays(3): Beta M CN
## rownames(786477): cg26928153 cg16269199 ... cg05111475 cg09226288
## rowData names(0):
## colnames(24): GSM4495491 GSM4495492 ... GSM4495513 GSM4495514
## colData names(6): Sample_Name Sample_Type ... Basename filenames
## Annotation
## array: IlluminaHumanMethylationEPIC
## annotation: ilm10b4.hg19
## Preprocessing
## Method: Illumina, bg.correct = TRUE, normalize = controls, reference = 1
## minfi version: 1.42.0
## Manifest version: 0.3.0
做完这些步骤后可以再次画图比较下:
par(mfrow=c(1,2))
# 原先的
mdsPlot(dat = RGset,
sampGroups = pd$Sample_Type,
sampNames = pd$Sample_Name
)
# 现在的
mdsPlot(dat = getBeta(mSetSqFlt),
sampNames = pd$Sample_Name,
sampGroups = pd$Sample_Type
)
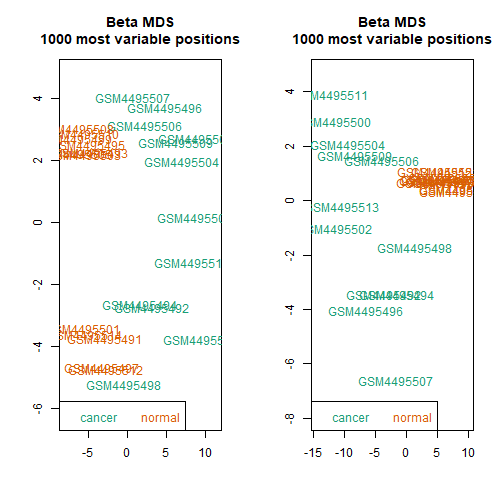
可以看到minfi的每个过滤步骤都是需要一步一步进行,在这方面ChAMP真的是太省心了,在读取数据时顺便就把这事干掉了!
后面就是差异分析了。差异分析需要用到M矩阵或者β矩阵,可以通过getM()/getBeta()函数从以上几个对象中提取。作者认为M矩阵具有更好的统计学特点,更适合统计分析,β矩阵更好解释生物学意义。
差异分析
也是分为3个层次:DMP,DMR,block
首先是DMP,我们这里用M矩阵进行差异分析:
myDMP.m <- dmpFinder(dat = getM(mSetSqFlt),
pheno = pd$Sample_Type,
type = "categorical", # 分组变量是分类变量
qCutoff = 1, # FDR q-value cutoff
shrinkVar = F # 样本数量小于10 用T
)
head(myDMP.m)
## intercept f pval qval
## cg22697045 -0.3495536 503.7669 1.180903e-16 3.936793e-11
## cg06319475 0.8345460 440.4474 4.857842e-16 8.097331e-11
## cg26199906 0.1397303 423.6200 7.309574e-16 8.122678e-11
## cg07774533 -0.9310782 411.6857 9.859642e-16 8.217309e-11
## cg26256223 1.2463769 401.0223 1.297355e-15 8.650023e-11
## cg16306898 0.4928779 385.1462 1.977991e-15 1.099010e-10
我们再用β矩阵试试看:
myDMP.b <- dmpFinder(dat = getBeta(mSetSqFlt),
pheno = pd$Sample_Type,
type = "categorical", # 分组变量是分类变量
qCutoff = 1, # FDR q-value cutoff
shrinkVar = F # 样本数量小于10 用T
)
head(myDMP.b)
## intercept f pval qval
## cg26256223 0.7010912 854.8678 4.219127e-19 1.414628e-13
## cg16601494 0.7142688 802.5722 8.304373e-19 1.414628e-13
## cg09296001 0.6612113 756.4304 1.565768e-18 1.778163e-13
## cg17301223 0.7292316 721.7549 2.586347e-18 2.202886e-13
## cg22697045 0.4400964 677.6529 5.070931e-18 3.250941e-13
## cg06319475 0.6382937 669.9864 5.725258e-18 3.250941e-13
取交集看看:
dmpM <- rownames(myDMP.m[myDMP.m$qval < 0.01,]) # 219677
dmpB <- rownames(myDMP.b[myDMP.b$qval < 0.01,]) # 190867
v1 <- VennDiagram::venn.diagram(x = list(dmpM=dmpM,dmpB=dmpB),
filename = NULL,
fill=c("#0073C2FF","#EFC000FF")
)
cowplot::plot_grid(v1)
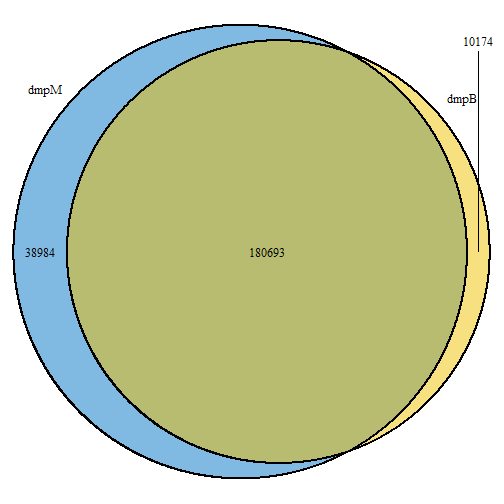
重合率还是非常高的!
然后是DMR:
design <- model.matrix(~pd$Sample_Type)
# 加速
library(doParallel)
registerDoParallel(cores = 6)
myDMR <- bumphunter(mSetSqFlt,
design = design,
coef = 2, # design的第2列
cutoff = 0.2,
nullMethod = "bootstrap",
type = "Beta"
)
## [bumphunterEngine] Parallelizing using 6 workers/cores (backend: doParallelSNOW, version: 1.0.17).
## [bumphunterEngine] Computing coefficients.
## [bumphunterEngine] Finding regions.
## [bumphunterEngine] Found 84939 bumps.
dim(myDMR$table)
## [1] 84939 10
head(myDMR$table)
## chr start end value area cluster indexStart
## 75901 chr10 118030848 118034031 -0.3772173 12.070953 55516 442352
## 78312 chr16 51183988 51186266 -0.3608858 11.548347 147315 629634
## 82985 chr6 29521013 29521803 -0.3570706 11.069188 326737 268459
## 75506 chr10 7450112 7453507 -0.4819137 10.120187 40434 415054
## 82452 chr5 127872767 127875163 -0.3819865 8.021716 312453 240183
## 81895 chr4 96469286 96471143 -0.3592760 7.904073 290890 202036
## indexEnd L clusterL
## 75901 442383 32 33
## 78312 629665 32 36
## 82985 268489 31 35
## 75506 415074 21 27
## 82452 240203 21 25
## 81895 202057 22 26
最后是block,这个网络上的教程非常少,最后还是查看了源码才知道怎么搞。
首先需要用cpgCollapse()函数得到一个cluster,然后就可以用blockFinder()计算差异block了。
cluster <- cpgCollapse(mSetSqFlt)
## [cpgCollapse] Creating annotation.
## [cpgCollapseAnnotation] Clustering islands and clusters of probes.
## [cpgCollapseAnnotation] Computing new annotation.
## [cpgCollapseAnnotation] Defining blocks.
## [cpgCollapse] Collapsing data
## ...........
## .............
myblock <- blockFinder(cluster$object,
design = design,
what = "Beta",
cutoff = 0.2,
nullMethod = "bootstrap")
## [bumphunterEngine] Parallelizing using 6 workers/cores (backend: doParallelSNOW, version: 1.0.17).
## [bumphunterEngine] Computing coefficients.
## [bumphunterEngine] Smoothing coefficients.
## [bumphunterEngine] Finding regions.
## [bumphunterEngine] Found 1404 bumps.
dim(myblock$table)
## [1] 1404 10
head(myblock$table)
## chr start end value area cluster indexStart indexEnd
## 206 chr2 217653964 218570889 0.2229784 38.35228 19 67800 67975
## 1248 chr15 60965059 61717754 0.2346791 25.81470 138 316674 316792
## 961 chr11 4936642 5510732 0.2422221 25.19110 112 244131 244234
## 541 chr5 157682514 158448438 0.2189335 24.95841 46 136911 137024
## 811 chr8 92568908 93738204 0.2232051 24.32935 76 198321 198437
## 789 chr8 68969455 70246427 0.2687594 22.84454 73 196345 196434
## L clusterL
## 206 172 6565
## 1248 110 6877
## 961 104 3462
## 541 114 9177
## 811 109 2305
## 789 85 2351
注释
可以直接通过getAnnotation()函数直接获得,还是很方便的,但是在ChAMP中是自动给你添加的,真的是考虑周到。
annoMset <- getAnnotation(mSetSqFlt)
dim(annoMset)
## [1] 786477 46
names(annoMset)
## [1] "chr"
## [2] "pos"
## [3] "strand"
## [4] "Name"
## [5] "AddressA"
## [6] "AddressB"
## [7] "ProbeSeqA"
## [8] "ProbeSeqB"
## [9] "Type"
## [10] "NextBase"
## [11] "Color"
## [12] "Probe_rs"
## [13] "Probe_maf"
## [14] "CpG_rs"
## [15] "CpG_maf"
## [16] "SBE_rs"
## [17] "SBE_maf"
## [18] "Islands_Name"
## [19] "Relation_to_Island"
## [20] "Forward_Sequence"
## [21] "SourceSeq"
## [22] "UCSC_RefGene_Name"
## [23] "UCSC_RefGene_Accession"
## [24] "UCSC_RefGene_Group"
## [25] "Phantom4_Enhancers"
## [26] "Phantom5_Enhancers"
## [27] "DMR"
## [28] "X450k_Enhancer"
## [29] "HMM_Island"
## [30] "Regulatory_Feature_Name"
## [31] "Regulatory_Feature_Group"
## [32] "GencodeBasicV12_NAME"
## [33] "GencodeBasicV12_Accession"
## [34] "GencodeBasicV12_Group"
## [35] "GencodeCompV12_NAME"
## [36] "GencodeCompV12_Accession"
## [37] "GencodeCompV12_Group"
## [38] "DNase_Hypersensitivity_NAME"
## [39] "DNase_Hypersensitivity_Evidence_Count"
## [40] "OpenChromatin_NAME"
## [41] "OpenChromatin_Evidence_Count"
## [42] "TFBS_NAME"
## [43] "TFBS_Evidence_Count"
## [44] "Methyl27_Loci"
## [45] "Methyl450_Loci"
## [46] "Random_Loci"
内容很全,染色体、起始位置、基因名字等都有了,有了这些东西就够了。
annoMset_subset <- annoMset[,c("chr",
"Name",
"Relation_to_Island",
"UCSC_RefGene_Name"
)]
head(annoMset_subset)
## DataFrame with 6 rows and 4 columns
## chr Name Relation_to_Island UCSC_RefGene_Name
## <character> <character> <character> <character>
## cg26928153 chr1 cg26928153 OpenSea DDX11L1
## cg16269199 chr1 cg16269199 OpenSea DDX11L1
## cg13869341 chr1 cg13869341 OpenSea WASH5P
## cg02404219 chr1 cg02404219 OpenSea OR4F5
## cg04098293 chr1 cg04098293 OpenSea
## cg16382250 chr1 cg16382250 OpenSea
简单!有了这些内容,后面的各种分析就水到渠成了!















 1万+
1万+











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









