






1.总体流程
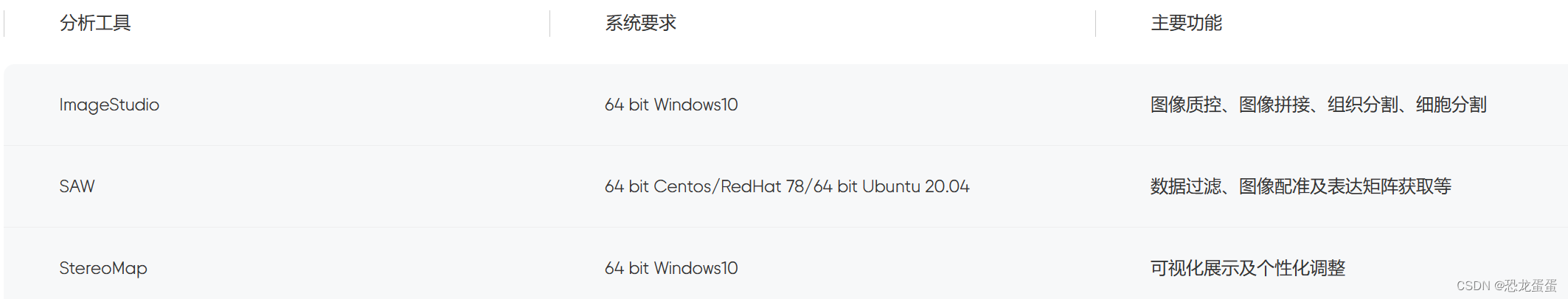


2.配置
在st环境中使用Singularity build的容器,但是好像没有安装Singularity ,执行命令就莫名其妙开始下载了
不过之前运行过 conda install singularity 和 conda install apptainer 记得出错了
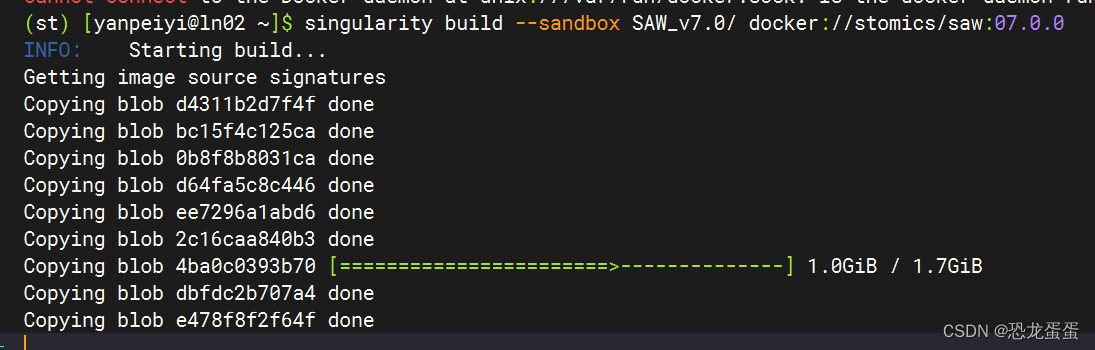
!找到问题了 但是其他依赖不安装也能build,先试试吧,不好使再说
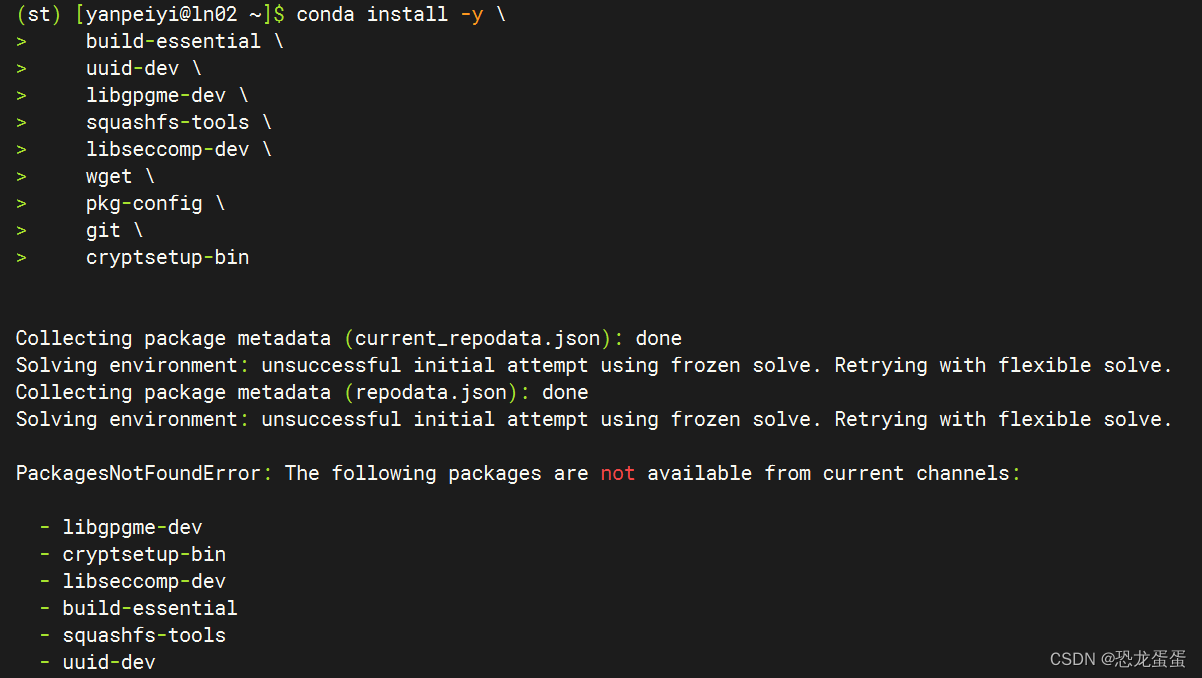
网络很慢 终于build好了
3.下载数据集
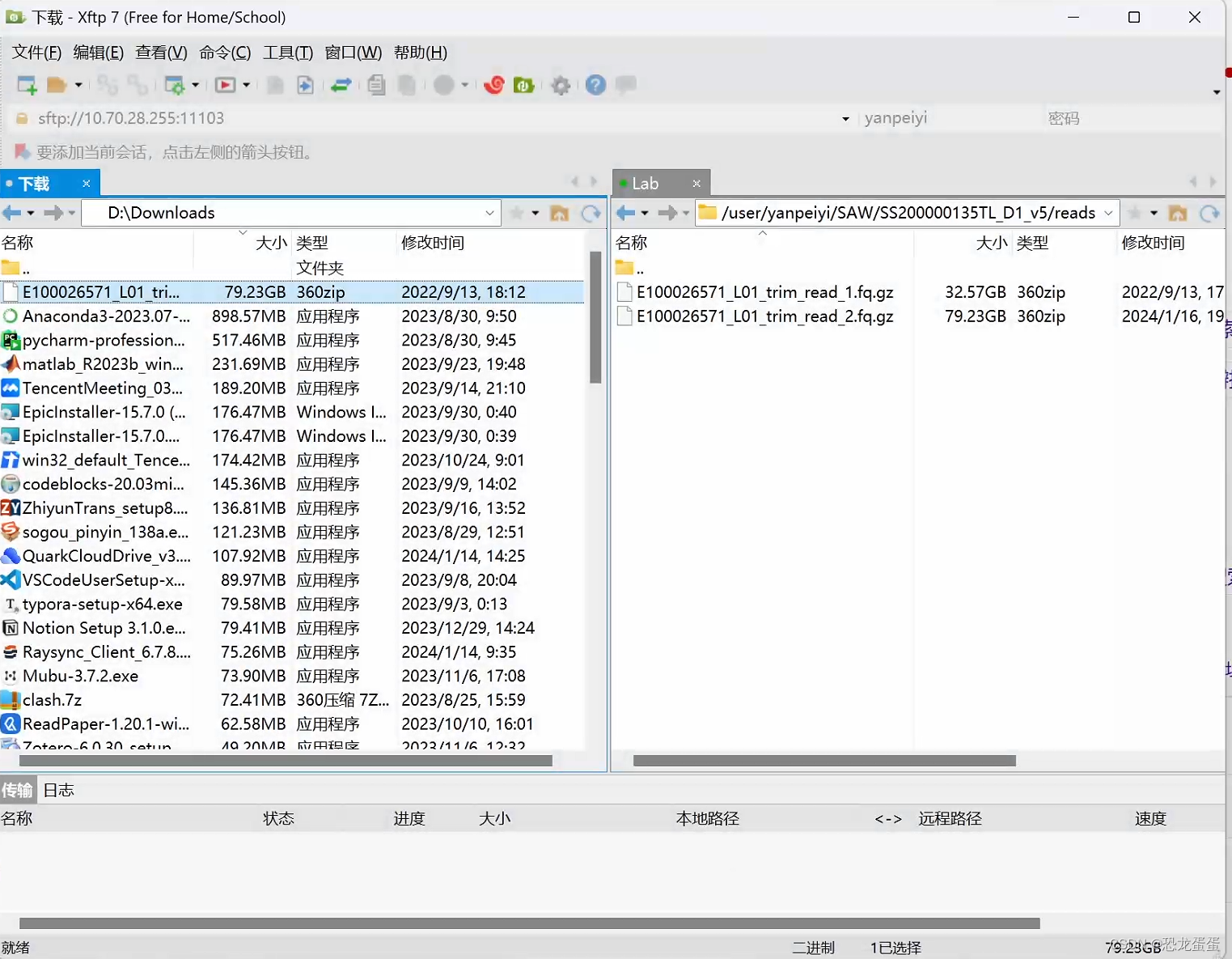
接下来开始下载数据集了,用的是v5的brain数据集
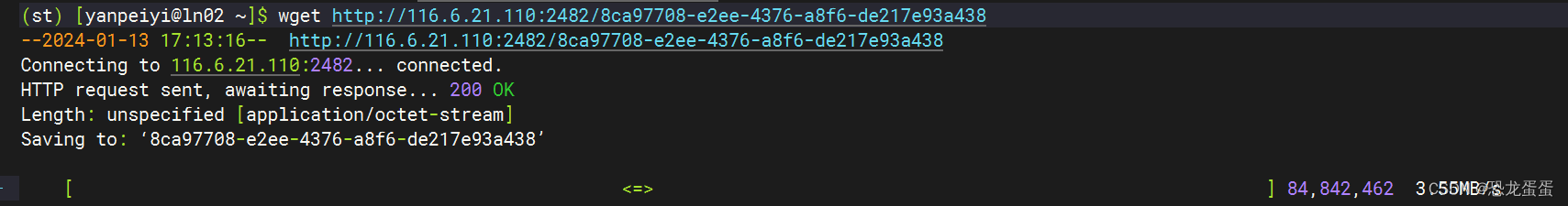
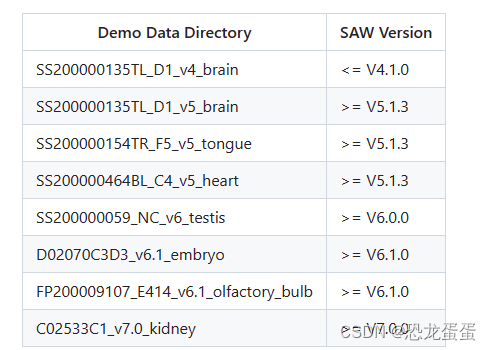
数据集下载老是暂停,因为是动态链接,怀疑是时间太久了,而且ssh连接昨晚断了一次,很奇怪。
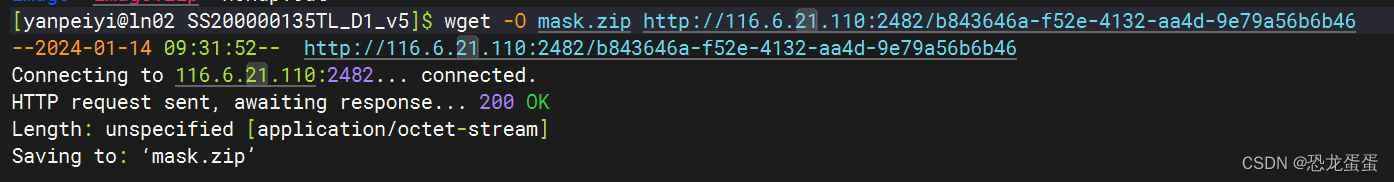
想了一个好方法:在服务器上下载很慢,因为校园网慢,所以在跳板机上连外网下载,再上传到服务器上,应该挺快。
下好了,在跳板机使用MobaXterm,但是80g的文件只能上传80%,没有找到原因
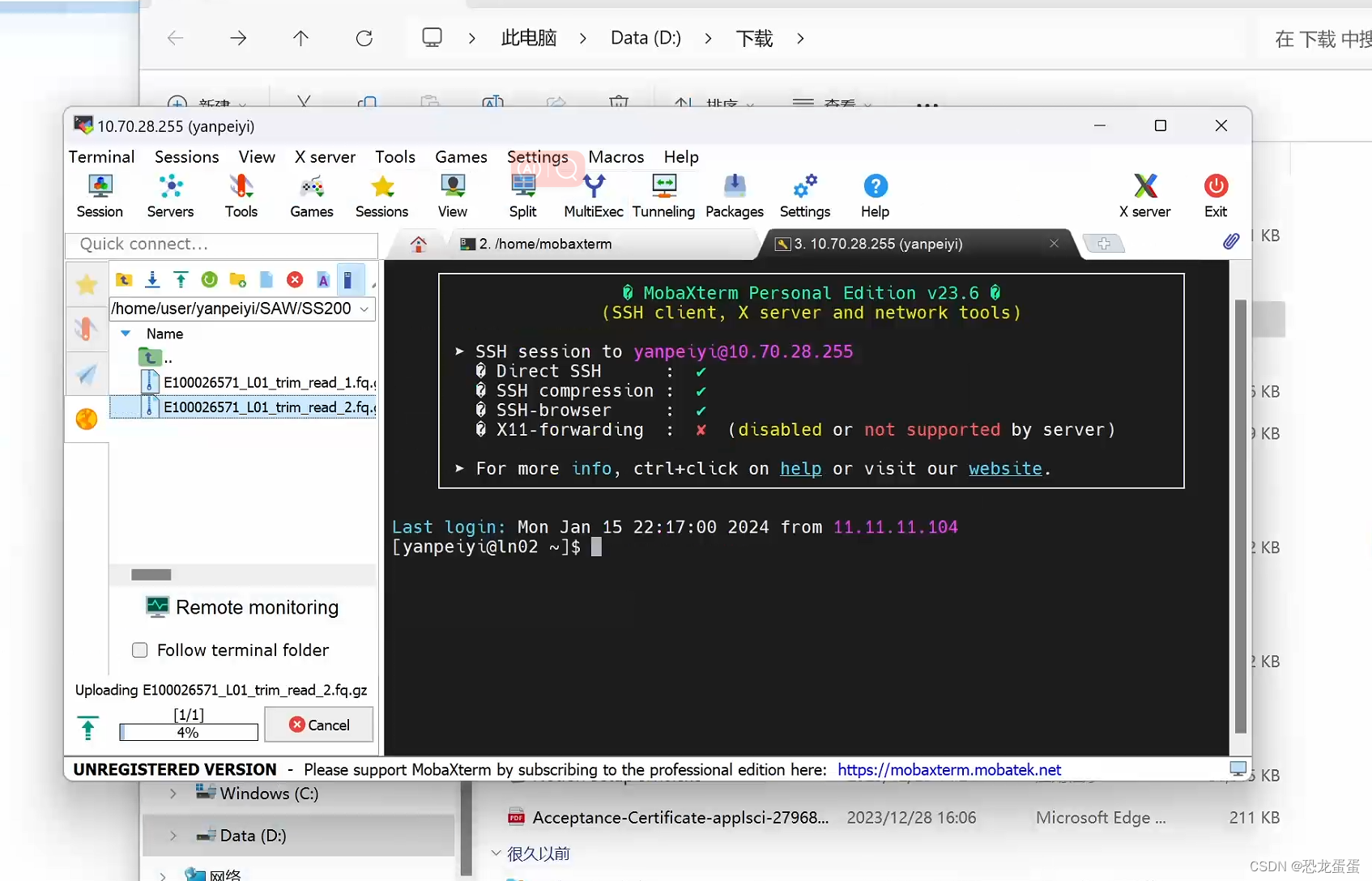
听了师兄的建议,使用xftp,顺利弄好数据集。果然还是要用稳定的app,花里胡哨的没用。
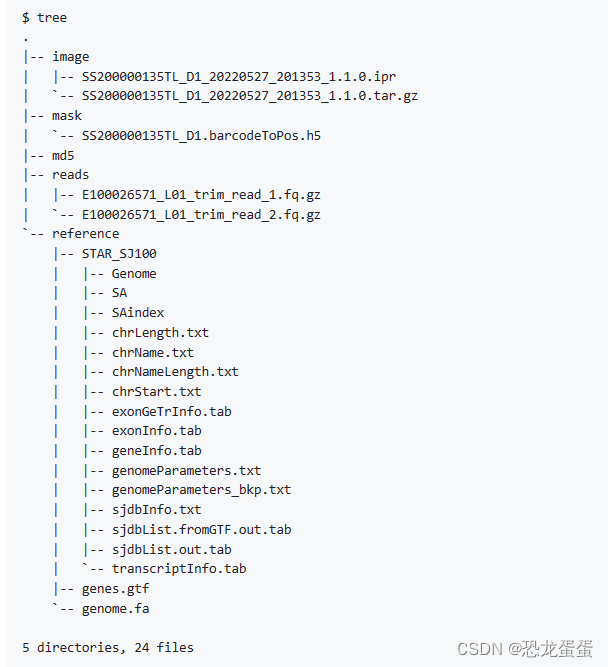
4.建立基因组索引
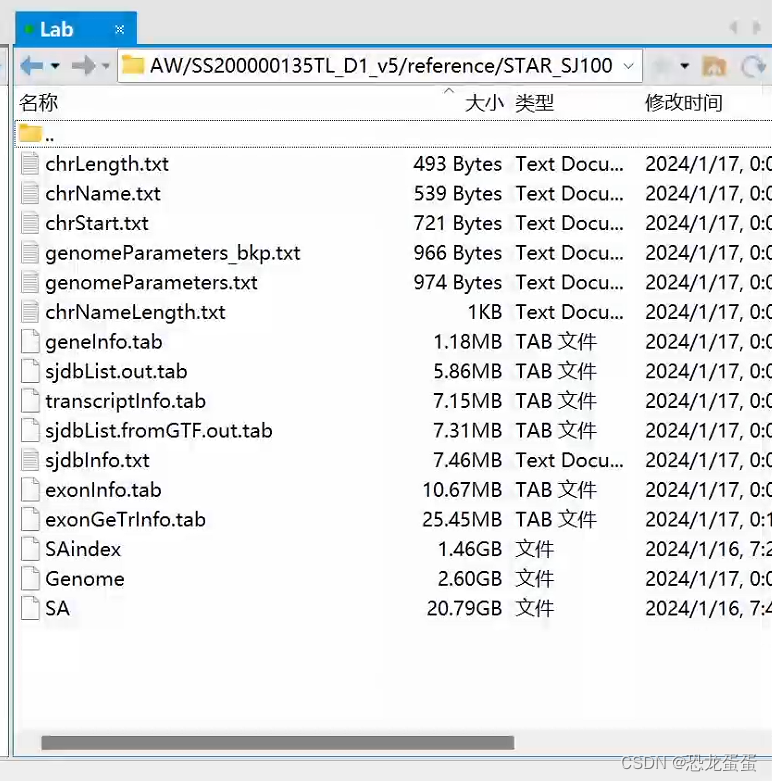
https://github.com/STOmics/SAW/tree/main/Scripts/pre_buildIndexedRef
referenceDir=/Full/Path/Of/Reference/Folder/Path
export SINGULARITY_BIND=$referenceDir
singularity exec <SAW_version.sif> mapping \
--runMode genomeGenerate \
--genomeDir $referenceDir/STAR_SJ100 \
--genomeFastaFiles $referenceDir/genome/genome.fa \
--sjdbGTFfile $referenceDir/genes/genes.gtf \
--sjdbOverhang 99 \
--runThreadN 12
Then you should get the mask file from our website through the slide number(SN)
5.split mask
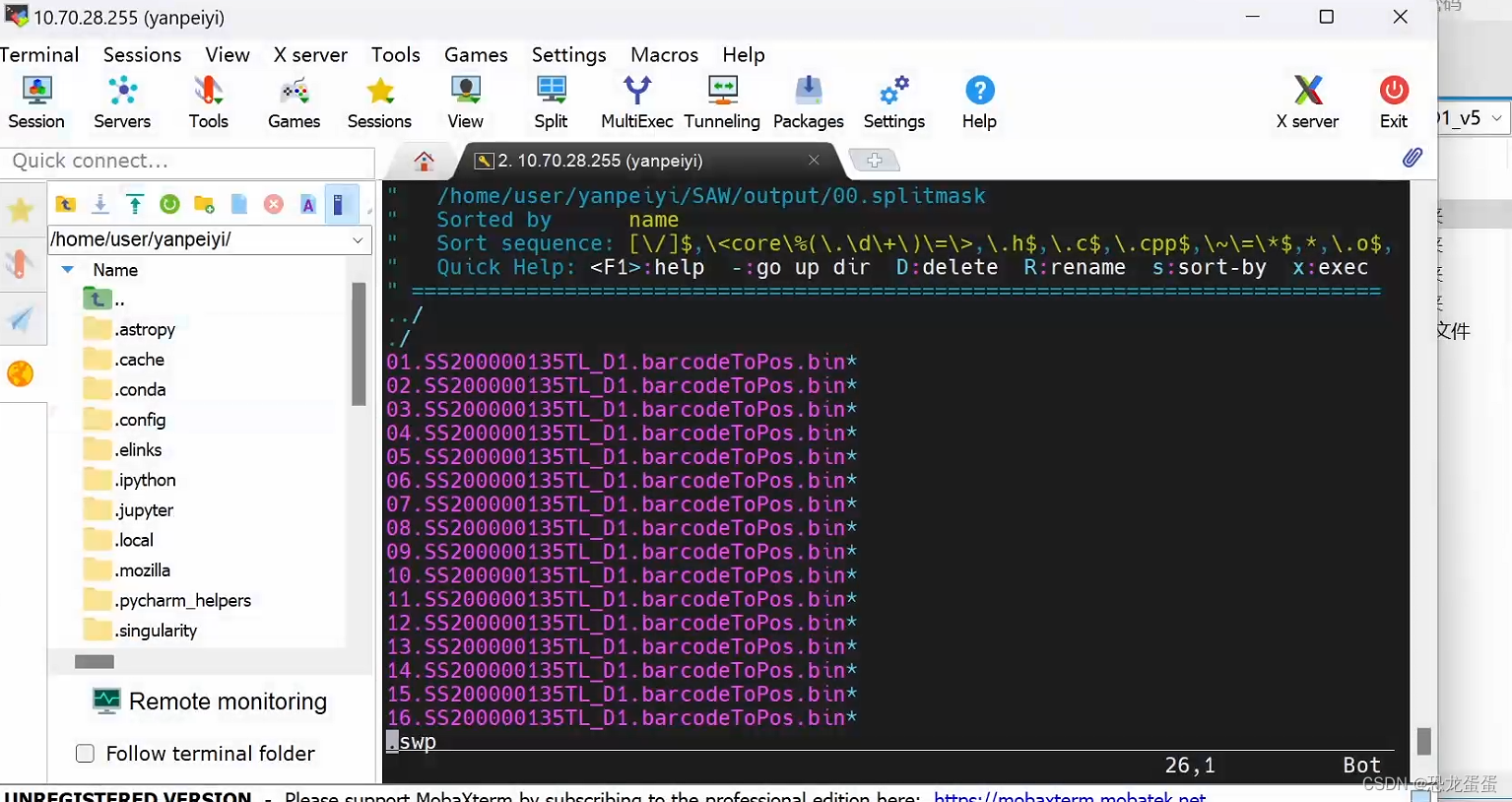
$ mkdir /path/to/output/00.splitmask
$ singularity exec SAW_v6.0.sif splitMask \
/path/to/data/{SN}.barcodeToPos.h5 \ # 必需参数:Stereo-seq 芯片 mask 文件(.h5)
/path/to/output/00.splitmask \ # 必需参数:输出目录
8 \ # 必需参数:运行线程数
16 \ # 可选参数:分割份数
2_25 # 可选参数
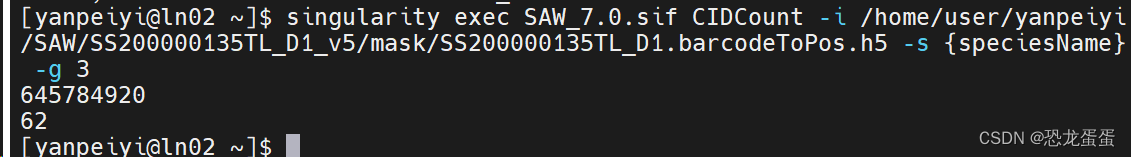
6.CIDCount:
是用于计算 Stereo-seq 芯片 mask 文件中 CID 数量,以及粗略估算 mapping 过程所需内存的小程序。
我没指定species 也行?
$ singularity exec SAW_v6.0.sif CIDCount \
-i /path/to/data/{SN}.barcodeToPos.h5 \ 必需参数:Stereo-seq 芯片 mask 文件(.h5)
-s {speciesName} \ 必需参数:物种名
-g {genomeSize} 必需参数 : 基因组大小
7.mapping:
每一条 Stereo-seq 序列包含一段 CID 序列,该序列是用于将测序 reads 比对在组织切片上的原始位置的关键信息。Stereo-seq 原始测序数据通过 SAW 软件中的 mapping 工具,将存储在 FASTQ 文件中的原始测序reads 的CID 与 Stereo-seq 芯片 Mask 文件记录的 CID 坐标键值对进行匹配(允许一位容错)。根据 mask 文件的记录为 CID 能够匹配的 reads 添加坐标信息。经过 CID 匹配的 reads 为具有有效 CID 的 mRNA reads (Valid CID Reads)。Valid CID Reads 经过滤后,得到 Clean Reads。mapping 工具将 Clean Reads 与相应物种的参考基组进行比对,并输出排序后的 BAM 格式比对文件和统计报告。
option 1 单对 PE FASTQ
option 2 多对 PE FASTQ
多对 PE FASTQ 和单对有啥区别
ChatGPT
多对 PE FASTQ 和单对的主要区别在于测序方式和数据处理。
多对 PE (Paired-End) FASTQ:
测序方式:在这种方法中,DNA或RNA片段的两端被同时测序。这意味着从同一个DNA片段生成两个读序,一个从一端开始,另一个从另一端开始。
数据处理:由于有两个读序,因此数据分析时可以更准确地拼接读序,特别是在重复序列或复杂基因组区域。这提供了对片段两端更好的覆盖,可以用于更准确的基因组重建和变异检测。
应用:多对 PE 测序在结构变异分析、基因组装配和转录组研究中特别有用。
单对 FASTQ:
测序方式:在单对测序中,只有DNA或RNA片段的一端被测序。这意味着每个DNA片段只生成一个读序。
数据处理:单对测序的数据处理比多对PE测序简单,但它提供的信息较少。它不能有效解决重复序列或复杂基因组区域的拼接问题。
应用:单对测序通常用于基因表达分析(如RNA-Seq)和某些基因组测序应用,尤其是当基因组较小或较简单时。
mid就是umi
$ mkdir /path/to/output/01.mapping ##option1(单对 PE FASTQ 新建目录)
$ mkdir /path/to/multi_lane_output/01.mapping ## option2(多对 PE FASTQ 新建目录)
$ vim /path/to/output/01.mapping/{lane}.bcPara ##option1(单对 PE FASTQ)创建参数配置
文件
$ vim /path/to/multi_lane_output/01.mapping/{lane}.bcPara ##option2(多对 PE FASTQ)
创建参数配置文件
in=/path/to/data/{SN}.barcodeToPos.h5 ## 必需参数:Stereo-seq 芯片 mask 文件(.h5)
in1=/path/to/data/{lane}_read_1.fq.gz ## 必需参数:Stereo-seq 原始测序数据 FASTQ read1
文件(.fq.gz)
in2=/path/to/data/{lane}_read_2.fq.gz ## 可选参数:Stereo-seq 原始测序数据 FASTQ read2
文件(.fq.gz)
encodeRule=ACTG ## 必需参数(PE 场景):碱基编码规则
out={lane} ## 可选参数:输出前缀
barcodeReadsCount=/path/to/output/01.mapping/{lane}.barcodeReadsCount.txt ## option1 (单对 PE FASTQ)必需参数:CID mapping 统计输出
barcodeReadsCount=/path/to/multi_lane_output/01.mapping/{lane}.barcodeReadsCount.
txt ## option2(多对 PE FASTQ)必需参数:CID mapping 统计输出
action=4 ## 必需参数:行动参数 (Valid options: 1=CID stat, 2=CID overlap, 3=get CID
position map, 4=map CID to slide, or 5=merge CID list)
platform=T10 ## 可选参数:测序平台A
barcodeStart=0 ## 必需参数:CID 起始位点
barcodeLen=25 ## 必需参数:CID 长度(PE 为 25)
umiStart=25 ## 必需参数:MID 起始位点
umiLen=10 ## 必需参数:MID 长度
umiRead=1 ## 必需参数:表明片段包含 MID
mismatch=1 ## 必需参数:mapping 的最大容错碱基数
bcNum=645784920 ## 必需参数:CID 数量,CIDCount 的输出文件第一行
polyAnum=15 ## 可选参数:polyA 长度
mismatchInPolyA=2 ## 可选参数:找寻 polyA 时最大容错碱基数
rRNAremove ## 可选参数:是否过滤 rRNA
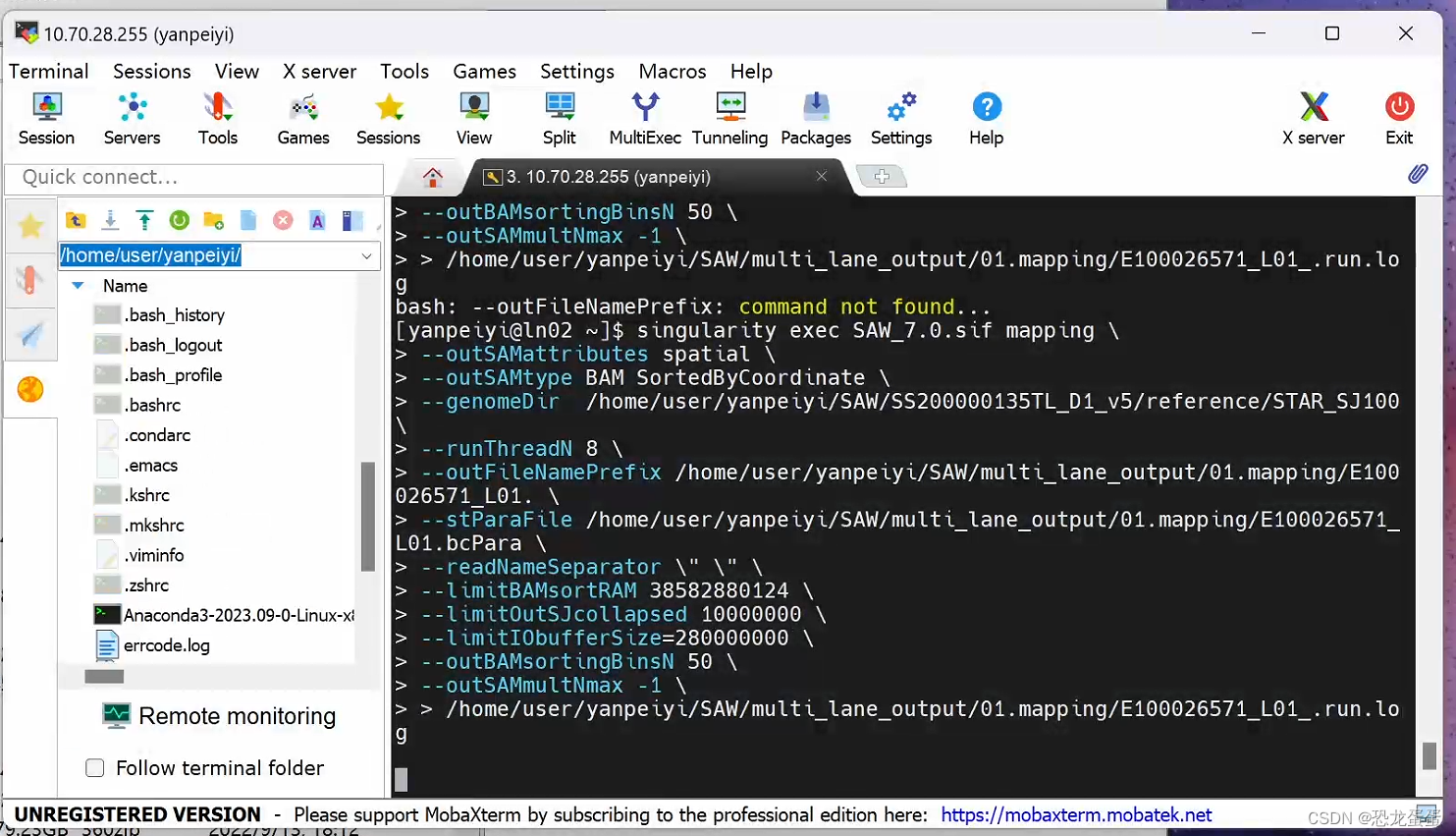
singularity exec SAW_7.0.sif mapping \
--outSAMattributes spatial \
--outSAMtype BAM SortedByCoordinate \
--genomeDir /home/user/yanpeiyi/SAW/SS200000135TL_D1_v5/reference/STAR_SJ100\
--runThreadN 8 \
--outFileNamePrefix /home/user/yanpeiyi/SAW/multi_lane_output/01.mapping/E100026571_L01. \
--stParaFile /home/user/yanpeiyi/SAW/multi_lane_output/01.mapping/E100026571_L01.bcPara \
--readNameSeparator \" \" \
--limitBAMsortRAM 38582880124 \
--limitOutSJcollapsed 10000000 \
--limitIObufferSize=280000000 \
--outBAMsortingBinsN 50 \
--outSAMmultNmax -1 \
> /home/user/yanpeiyi/SAW/multi_lane_output/01.mapping/E100026571_L01_.run.log
下图我用的 multi lane的方式,应该也可以one lane的方式mapping再合并,但是我不确定前者可以用

















 6056
6056

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









