






 CheckM2是一种采用通用和特定机器学习模型评估基因组bin质量和污染情况的工具。它能处理不同分类学谱系,对稀有或特殊生物体提供高精度预测。文章介绍了安装、数据库下载、使用方法及输出解读,适用于宏基因组和扩增子分析。
CheckM2是一种采用通用和特定机器学习模型评估基因组bin质量和污染情况的工具。它能处理不同分类学谱系,对稀有或特殊生物体提供高精度预测。文章介绍了安装、数据库下载、使用方法及输出解读,适用于宏基因组和扩增子分析。

简介
CheckM2使用机器学习快速评估基因组bin质量
与CheckM1不同,CheckM2采用通用训练的机器学习模型,无论分类学谱系如何,均可用于预测基因组bin的完整性和污染情况。这使得它能够在训练集中纳入许多仅具有少数(甚至只有一个)高质量基因组代表的谱系,通过将其置于训练集中所有其他生物体的背景下进行分析。得益于这一机器学习框架,CheckM2对于具有缩减基因组或特殊生物学特性的生物体,如Nanoarchaeota或Patescibacteria,也具有极高的准确性。
CheckM2使用两种独立的机器学习模型来预测基因组完整性。其中,“通用”梯度提升模型具有良好的泛化能力,适用于GenBank或RefSeq中代表性不足的生物体(大致相当于在目、纲或门水平上为新物种)。而“特定”神经网络模型在预测与参考训练集相近物种的完整性时更为准确(大致相当于已知物种、属或科中的生物体)。CheckM2通过计算余弦相似度自动确定适用于每个输入基因组的完整性模型,但用户也可以强制使用特定的完整性模型,或者获取两种模型的预测输出。至于污染情况,CheckM2只有一种基于梯度提升的模型,该模型不受不同版本分类学物种注释的影响,适用于所有情况。
安装
mamba env create -f checkm2.yaml
mamba activate checkm2
checkm2.yaml文件如下
names: checkm2
channels:
- conda-forge
- bioconda
- defaults
dependencies:
- checkm2=1.0.1
下载数据库
官方下载链接
https://zenodo.org/api/files/fd3bc532-cd84-4907-b078-2e05a1e46803/checkm2_database.tar.gz
或者从我们的网盘中下载
链接:https://pan.quark.cn/s/e714d2d9d7f2
提取码:u2R1
解压
tar -xf checkm2*z
使用
checkm2 predict \
--threads 16 \
--input ./Bin/ \
--output-directory ./Bin_quality/ \
--database_path ./checkm2/uniref100.KO.1.dmnd
--input输入目录,包含bin.fa或bin.fna等文件
Bin.1.fa
Bin.2.fa
Bin.3.fa
Bin.4.fa
Bin.5.fa
Bin.6.fa
...
--output-directory输出文件目录--database_path数据库文件--threads所使用的线程数
输出结果
- diamond_output/
- protein_files/
- checkm2.log
quality_report.tsv
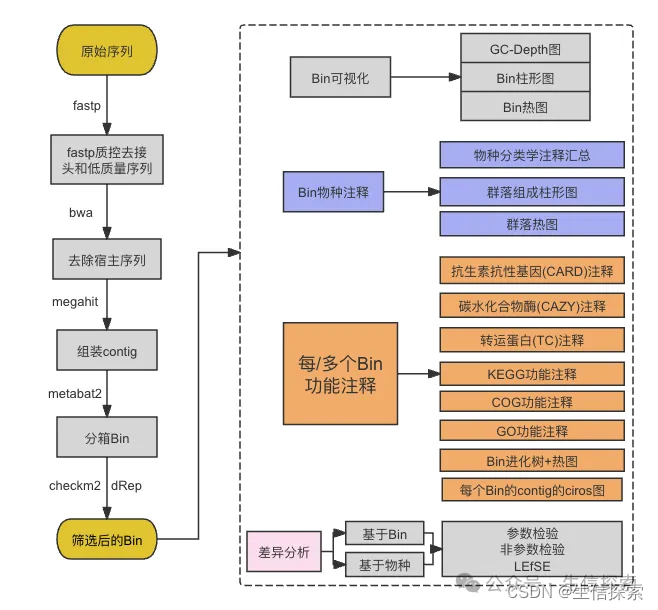
可以根据quality_report.tsv文件中的Completeness>75和Contamination<10挑选Bin
grep 'Bin' Bin_quality/quality_report.tsv | \
awk '{if($2>75 && $3<10) print $1}' > Bin_quality/checkm2_pick.txt
| Name | Completeness | Contamination | Completeness_Model_Used | Translation_Table_Used | Coding_Density | Contig_N50 | Average_Gene_Length | Genome_Size | GC_Content | Total_Coding_Sequences | Additional_Notes |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Bin.31 | 64.52 | 4.61 | Gradient Boost (General Model) | 11 | 0.881 | 4705 | 287.0520945 | 1818772 | 0.63 | 1862 | None |
| Bin.310 | 87.92 | 2.31 | Neural Network (Specific Model) | 11 | 0.841 | 19758 | 357.3994819 | 2456844 | 0.51 | 1930 | None |
| Bin.311 | 22.07 | 0.01 | Neural Network (Specific Model) | 11 | 0.852 | 16495 | 304.8594595 | 594476 | 0.42 | 555 | None |
| Bin.312 | 70.1 | 0.93 | Neural Network (Specific Model) | 11 | 0.868 | 4467 | 273.7622999 | 1592766 | 0.38 | 1687 | None |
| Bin.313 | 5.45 | 0.02 | Neural Network (Specific Model) | 11 | 0.925 | 62916 | 244.9387755 | 232156 | 0.41 | 294 | None |
| Bin.314 | 91.29 | 0.5 | Gradient Boost (General Model) | 11 | 0.954 | 14158 | 348.0688497 | 1299713 | 0.49 | 1191 | None |
Reference
https://github.com/chklovski/CheckM2
https://pubmed.ncbi.nlm.nih.gov/37500759/
承接宏基因组、扩增子全部分析内容


















 365
365

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









