











发表期刊:Computational and Structural Biotechnology Journal(IF=7.271)
发表时间:2021
研究背景
微生物是生物和环境的营养循环和代谢过程所必需的,并且与生理学至关重要。虽然微生物可以使用传统培养的方法来确定,但目前自然界中可培养的微生物仅为1%。宏基因组能够通过测序从混合DNA中检测不可培养的微生物基因组序列(图1A),通过binning方法,将数据中来自同一菌株的序列聚到一起,得到单个菌株的基因组。
2021年底,发表在《Computational and Structural Biotechnology Journal》期刊的这篇综述详细阐述了宏基因组binning分析中用到的各类工具,给广大生信分析者提供了重要参考。
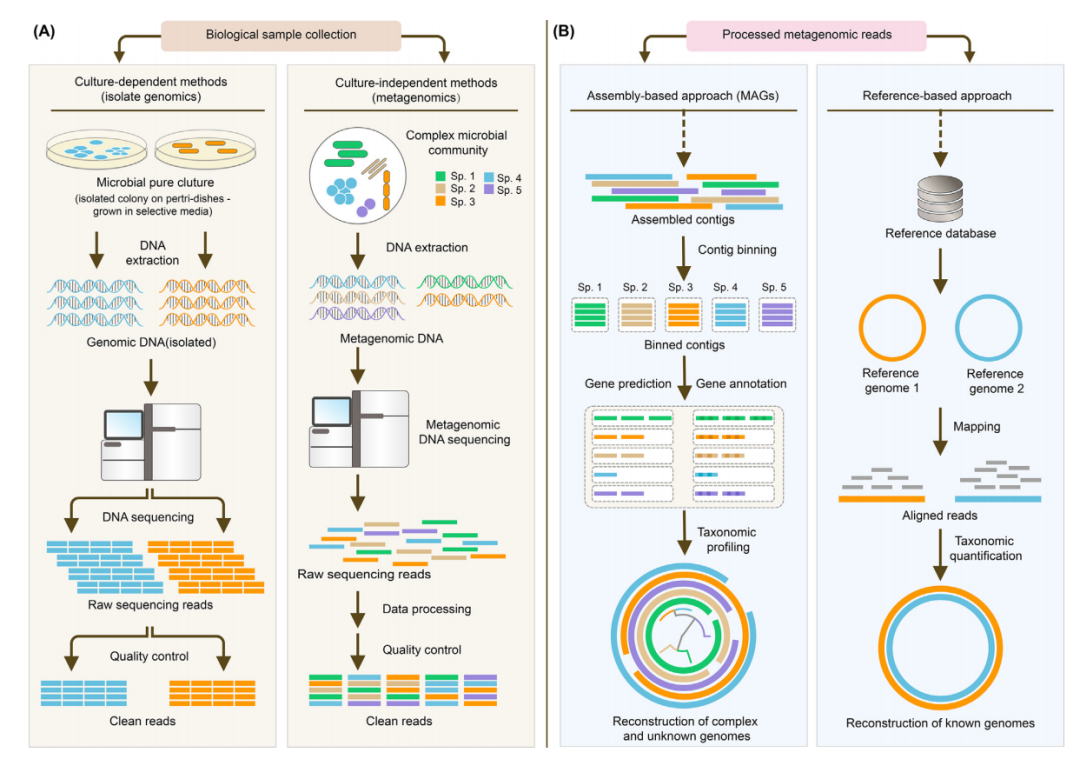
图1 在宏基因组学研究领域中使用的不同方法的示意图表示
(A)非培养(宏基因组学)方法和依赖于培养的方法之间的示意图对比。(B)基于组装和基于参考数据库方法的宏基因组测序数据之间的对比。
宏基因组项目分析工具
1、序列质控工具
针对短读长测序(如Illumina测序),常用的序列质控工具有FastQC,fastp、Trimmomatic和SOAPnuke(表1)。FastQC提供了碱基质量、GC含量分布和核苷酸偏倚的可视化说明。Fastp通过使用高级的多线程实现,提高了QC处理速度。Trimmomatic和SOAPnuke是分别针对Illumina和BGISEQ测序平台进行优化的最常用的两种工具。
针对长读长测序(如PacBio、Nanopore测序等),由于质控原理不同,相应的工具也有所不同,如SequelTools可通过过滤低质量reads并产生多个统计图,来检查PacBio长读长的质量。
表1 用于序列质控的工具

2、宏基因组组装工具
针对不同测序数据类型,宏基因组组装工具有所不同。传统短读长测序的宏基因组组装工具是基于OLC(overlap-layout consensus)方法设计的,如Omega。还有其他几种使用DBG(De Bruijn graph)设计的工具,如MetaVelvet、MetaVelvet-SL和MEGAHIT。
针对SLR(synthetic long reads)和linked reads测序,分别有Nanoscope、Athena和cloudSPAdes工具。由于PacBio和ONT平台生成的长reads比二代NGS测序具有更高的碱基错误率,针对长读长已经开发了用于碱基错误修正的专用模块,例如Canu和NECAT,使用OLC方法纠正了基因组组装前长序列的测序错误。
短读长和长读长测序技术在某种程度上是互补的,因为短读长具有较高的碱基质量,而长读长提供了连通性。因此,一些算法通过利用两种测序技术的优点开发了新的工具,如DBG2OLC和Opera-MS。
总之,二代宏基因组组装最常用的工具是MEGAHIT,三代宏基因组组装常用是MetaFlye和Canu。
表2 宏基因组组装工具

3、宏基因组组装结果检验工具
有许多工具可用来评估宏基因组组装生成的contigs和scaffolds的准确性和连续性。
● MetaQUAST可以快速计算出连续长度和scaffolds的基本统计数据,如组装长度、N50值和连续长度分布等。
● REAPR可以精确地识别基因组组装中的错误,且不依赖于参考序列。
● VALET在QC之前进行宏基因组binning,以减少由于reads深度不均匀而导致的假阳性和假阴性的数量。
● DeepMAsED可不依赖参考基因组,使用深度学习模型来检测错误组装的序列。
表3 用于组装结果检验的工具

4、宏基因组binning工具
目前的大多数组装工具并不能以单一scaffolds代表完整的微生物基因组。许多宏基因组binning工具被开发出来,将scaffolds分成簇,以代表一个生物体的整个基因组(表4)。Metabat2在众多单样本分箱工具中具有很大优势,从Bin数量、完成度、Bin纯度、精确度、效率上来看,Metabat2表现都十分出色。
表4 宏基因组binning的工具

5、MAGs完整度和污染度评估
CheckM通常用于确定每个bins的质量。然后只选择质量相对较高的bins作为后续注释的MAG。根据其完整性、污染水平和rRNA/tRNA预测,这些bins通常分为高质量、中等质量和其他类型。
表5 MAGs质量评估
| 类型 | 完整度 | 污染度 |
| 高质量MAGs | >90% | <5% |
| 中等质量MAGs | ≧50% | <10% |
| 其他 | <50% | ≧10% |
6、基因预测工具
隐马尔可夫模型(HMM)是基因预测中最常用的算法。常用工具包括MetaGeneMark,Glimmer-MG和FragGeneScan。
一些针对细菌和古细菌基因组的基因预测工具,使用的是动态编程,例如Prodigal、MetaGen和MetaGeneAnnotator。
此外,各种深度学习工具在基因预测方面得到了相当多的关注,常用的工具有Meta-MFDL和CNN-MGP。
表6 基因预测工具

7、基因功能注释工具
宏基因组基因功能注释工具可分为两类:
1)宽泛功能的工具来评估完整的功能潜力;
2)特定功能的工具,专注于一个或几个特定的生物过程。
基于同源性的工具通常依赖于BLAST来比较预测基因序列与已知基因序列的相似性,这些工具在处理从MAG中预测出的大量基因时通常非常缓慢。而eggNOG-mapper、GhostKOALA、MG-RAST和PANNZER2,采用了优化的比对策略,使基因序列与数据库的比对速度提高100-1000倍。
表7 基因功能注释工具

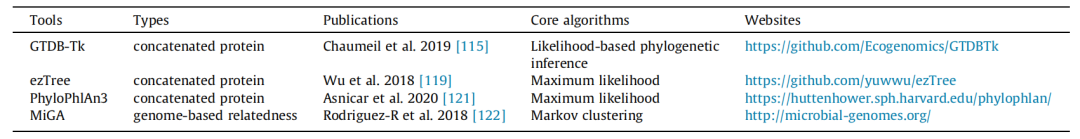
8、用于MAGs分类学鉴定的工具
注释MAGs时的另一个关键任务是确定它们的分类学地位。传统的基于16S rRNA的分类方法分辨率有限,在MAGs中的代表性较差。相比之下,单拷贝标记基因由于其分辨率的提高而受到欢迎(表7)。
推荐GTDB-Tk软件工具包,该工具基于基因组分类数据库GTDB可以对宏基因组binning获得的单菌基因组(MAGs)进行分类鉴定。
表8 MAGs分类学鉴定工具
9、分析MAGs丰度工具
用来估计宏基因组测序数据中MAGs丰度的具被分为四类:
1)基于蛋白质的工具;
2)基于k-mer的工具;
3)基于标记基因的工具;
4)基于单核苷酸多态性(SNP)的工具。
这四种方法都能估计MAGs丰度,但具有不同的分辨率。例如,基于k-mer的工具计算了MAGs的特定序列的丰度,而基于标记基因的工具计算了分类学丰度。
表9 用于分析MAG丰度的工具

总结
本研究为宏基因组binning上游和下游的分析工具提供了统一的公共资源,读者可以根据自己的研究目标选择最有效的工具和软件应用程序,同时文章内容加速了在宏基因组学领域使用的相关软件、工具等的研究和开发进程。
参考文献
A review of computational tools for generating metagenome-assembled genomes from metagenomic sequencing data. 2021.
DOI:10.1016/j.csbj.2021.11.028
















 8944
8944











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











