







Forcite Plus 是一款分子力学和分子动力学模拟程序。它可以对分子、表面或三维周期性材料体系进行快速的能量计算、几何优化以及各种热力学条件下的动力学模拟研究,可以分析材料体系的各种结构参数、热力学性质、力学性质、动力学性质以及统计学性质。主要应用于有机、无机小分子、有机金属络合物、 高分子聚合物、纳米及多孔材料、部分金属、金属氧化物晶体及晶体表界面结构的研究。
Forcite Plus的主要功能
能量计算 吸附热,表面能等结构优化 优化原子坐标和晶胞参数,支持原子笛卡尔坐标和晶胞参数的限定,可以添加外应力(等静压)模拟淬火 将动力学模拟和结构优化相结合,辅助扫描势能面,寻找最优的分子构象、吸附构象等 。
模拟退火 基于不同温度点的动力学模拟,实现体系的反复升、降温过程,辅助扫描势能面,寻找最优的分子构象、吸附构象等
水分子与云母(mica_2d)建模优化过程
首先在晶体库中导入云母分子并且利用Amorphous Cell模块建立密度为1g/cm3的水分子晶格常数和云母分子设为一致,以便之后进行接合建模。
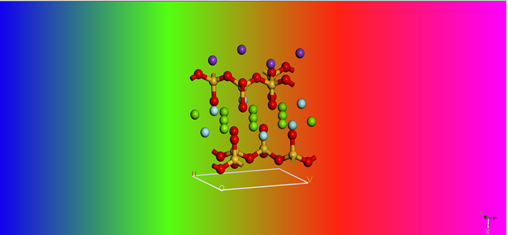
mica_2d
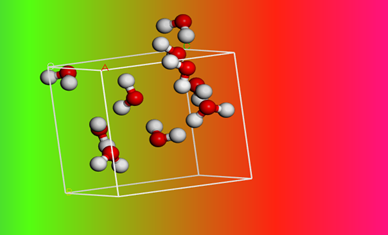
 H2O
H2O
Amorphous Cell Calculation 设置参数如下
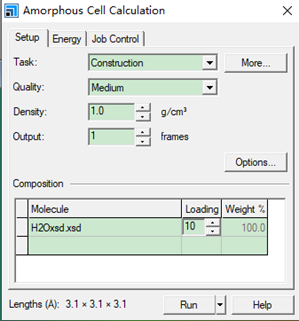
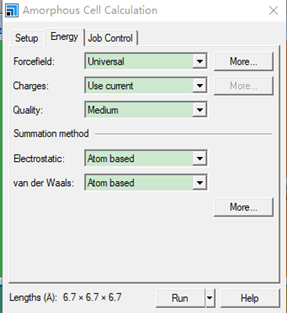
建模如下
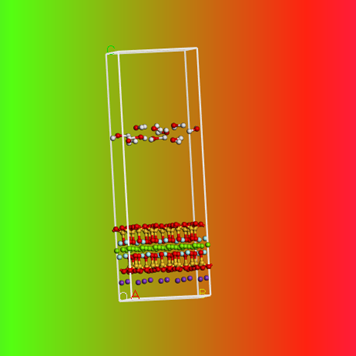
下面利用Forcite模块进行分子动力学模拟
以Forcite模块中的几何优化( GeometryOptimization)Smart方法来消除原子间距离过近或是重叠等情况,生成动力学模拟的初始构型。之后在Dynamics下进行分子动力学模拟,NVT系综,温度设为298.0 K,Universial力场,Noose-HoverLangevin控温,静电作用使用Ewald方法,van der Waals力则以Atom based 求解,截断半径cutoff为12.5A。
动力学模拟结果与初始结构对比
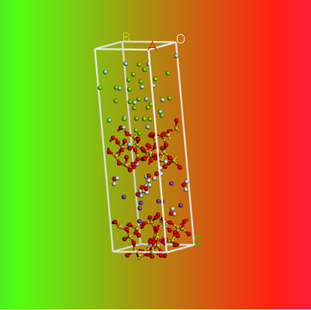
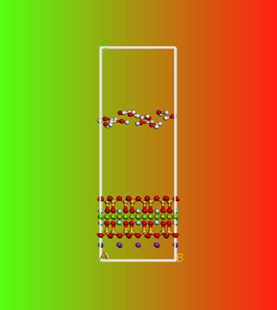
最后,有分子动力学模拟相关需求或者想加入我们都可以通过微信公众号联系我们。
微信公众号:320科技工作室


















 5272
5272

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











