










Multi-omics provide insights into the regulation of DNA methylation in pear fruit metabolism
多组学研究揭示梨果实代谢中DNA甲基化的调控机制

摘要
背景 尽管作物果实发育的研究较为深入,但多年生果树代谢调控网络的研究仍然有限。为填补这一知识空白,本研究对梨果实果肉在11个发育阶段(从幼果到成熟)的代谢组、蛋白质组、转录组、DNA甲基化组和小RNA组进行了全面分析,系统探讨了代谢格局和相关调控网络。
结果 我们构建了一个由439种代谢物和14,399个基因组成的关联数据库,以阐明梨果肉代谢的基因调控网络。令人惊讶的是,在梨果肉发育过程中,大多数数据库基因的启动子区DNA甲基化水平显著升高。通过对发育中的果实施用DNA甲基化抑制剂,发现其可抑制果皮中的叶绿素降解,同时促进果肉中叶黄素、β-胡萝卜素和脱落酸(ABA)的积累。此外,研究表明,梨果肉发育过程中ABA的逐步增加与类胡萝卜素代谢通路基因及多种转录因子的表达密切相关。其中,锌指蛋白PbZFP1被鉴定为梨果肉ABA生物合成的正向调节因子。尽管大多数ABA通路基因和转录因子的启动子区受到DNA甲基化修饰,但某些基因在DNA甲基化抑制剂的作用下得以诱导表达。结果表明,DNA甲基化通过抑制ABA积累可能延缓果实成熟。
结论 我们的研究揭示了梨果肉发育过程中代谢调控网络的表观遗传调控机制,特别是DNA甲基化的作用。
引言
物生命周期中的独特过程,为人类提供了富含健康促进成分的代谢物。在果实代谢物积累的同时,果实扩展和成熟伴随着一系列生理变化。目前在番茄果实中已鉴定出7118个代谢峰,但仅有22%被注释[1]。其中362种代谢物可通过广泛靶向代谢组学方法轻松检测到[2]。类似地,在桃和橙果实中检测到超过2000个代谢峰,但仅有493种代谢物被注释[3,4,5]。这些代谢物分类包括糖、有机酸、类黄酮、脂肪酸、多酚、萜类和植物激素等[6,7,8]。尽管这些代谢物的生物合成途径在不同物种间相似,但其时空分布和自然变异在不同物种乃至同一物种的不同品种间差异显著[4]。
代谢通路由一系列酶催化,这些酶可通过蛋白质组测序在肉质果实中部分鉴定[9,10]。然而,仅基于蛋白质丰度研究果实代谢通路的调控网络具有挑战性[11-16]。相比之下,转录组分析更常用于探索果实中的代谢通路。例如,在番茄果实中,转录组和代谢组数据的整合揭示了多酚类、甾醇糖苷和类黄酮代谢的调控网络[17,18]。此外,SlMYB12转录因子激活的基因-代谢物网络调控着粉色到红色色素的转化[2]。
转录活性受染色质结构的影响,而染色质结构由DNA甲基化和组蛋白的翻译后修饰(PTMH)调控。与PTMH相比,DNA甲基化在肉质果实中研究更为广泛,与花青素积累[19,20]、风味代谢[21,22]以及果实发育与成熟[23-26]密切相关。在植物中,DNA甲基化发生于对称的CG和CHG位点,以及不对称的CHH位点(H代表A、T或C)。在拟南芥中,CG、CHG和CHH甲基化分别由MET1、CMT3和CMT2维持,而通过RNA介导的DNA甲基化(RdDM)途径由DRM建立[27-31]。
尽管拟南芥和部分肉质果实中DNA甲基化机制已被阐明,但其在果实发育中的调控作用仍知之甚少。梨是全球栽培的水果之一,通过甲基化敏感扩增多态性、甲基化特异性内切酶PCR、薄层色谱检测以及全基因组亚硫酸盐测序等方法对其DNA甲基化进行了研究[36-40]。研究表明,DNA甲基化在秋水仙碱诱导的多倍体和长期储存的干燥种子中增加[36,39]。在梨悬浮培养细胞中,DNA甲基化抑制剂5-氮杂胞苷(5’-Aza)可降低乙烯的生成[41]。此外,MYB10和GA2ox8启动子中的胞嘧啶甲基化影响果皮花青素积累[19,37,38]。然而,DNA甲基化对梨果实代谢的影响仍不清楚。
在本研究中,我们对梨果实果肉发育的11个阶段进行了代谢组、转录组、蛋白质组、DNA甲基化组和小RNA组的分析。多组学数据的整合分析揭示了梨果肉发育过程中DNA甲基化和脱落酸(ABA)含量的全局升高。应用5’-Aza可显著提高ABA生物合成基因及其上游转录因子的表达,导致果肉中ABA的显著积累。这些新发现为理解DNA甲基化在梨果肉代谢物生成中的调控作用提供了新视角。
结果
梨果肉发育中的代谢谱分析
为了研究梨果肉发育过程中代谢物的变化,首先以‘砀山酥梨’(cv. Dangshansuli)果实为研究对象,分别在花后4周(S1)、6周(S2)、8周(S3)、10周(S4)、12周(S5)、14周(S6)、16周(S7)、18周(S8)、20周(S9)、22周(S10)和24周(S11)收集样本(图1a)。在此期间,观察到果实发育的典型特征(附加文件1:图S1),包括单果重、纵横径和可溶性糖含量的逐渐增加。在S10和S11阶段,乙烯含量显著增加;从S1到S3,石细胞含量增加,但随后逐渐减少;从S8到S11,果肉硬度逐渐下降,而可溶性固形物含量从S4到S11逐渐增加。
其次,代谢测定共鉴定出梨果肉中的492种代谢物。每个发育阶段平均检测到约456种代谢物,其中371种代谢物在所有阶段中普遍存在(附加文件2:表S1)。主成分分析(PCA)显示果肉在11个发育阶段之间存在显著差异(图1b)。通过比较不同发育阶段的相对含量(附加文件2:表S2),共鉴定出449种差异积累代谢物(DAMs)。这些DAMs属于32种不同类别,包括植物激素、花青素、氨基酸及其衍生物、碳水化合物、黄酮、黄酮醇、羟基肉桂酸衍生物、脂类、核苷酸及其衍生物、有机酸等。根据积累趋势,这些DAMs被分为八个簇(I→VIII)(图1c,附加文件2:表S3)。
-
簇I、II和IV:这些代谢物在早期果肉发育阶段(如S1至S3)水平较高(附加文件1:图S2),表明其与果肉的早期发育有关。例如,簇II中的丁香醇与果实木质化有关,该过程在花后15至55天的梨果肉中较为丰富[42]。
-
簇VII和VIII:这些代谢物在S7和S6阶段的果肉中水平较高(附加文件1:图S2),表明其可能与果实膨大相关。例如,簇VII中的L-丙氨酸、L-苯丙氨酸、L-脯氨酸和L-缬氨酸可能促进果实膨大[43, 44]。已有研究表明,氨基酸在梨果实膨大过程中显著积累[45]。
-
簇III、V和VI:这些代谢物在果实成熟阶段(S10和S11)水平较高(附加文件1:图S2),表明其可能与果实成熟相关。例如,簇V中的脱落酸(ABA)可以增加可溶性糖(包括蔗糖和葡萄糖)的含量,并促进乙烯释放,从而加速梨果实成熟[46]。
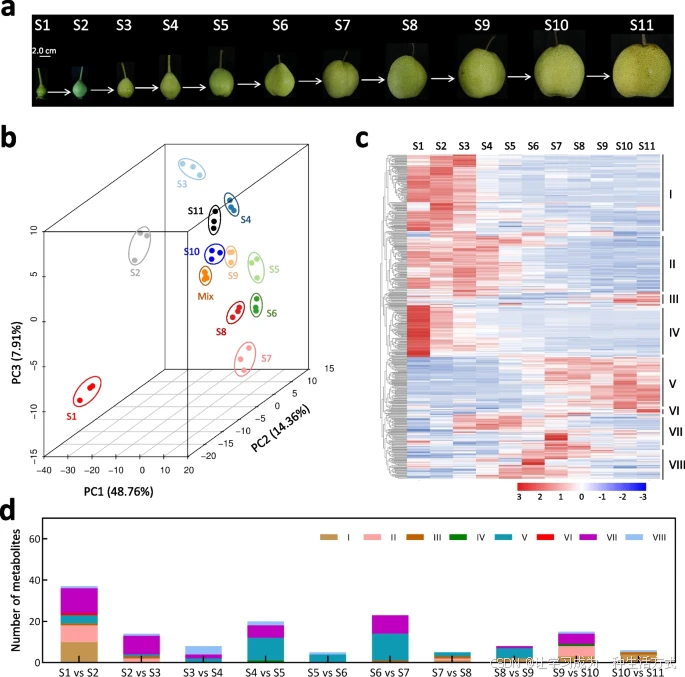
梨果肉发育过程中代谢物的动态变化
a 梨果实在11个发育阶段的采集样本。 b 对果肉在11个阶段的代谢组数据进行主成分分析(PCA)。 c 差异积累代谢物(DAMs)的聚类分析,将代谢物分为八个簇(I → VIII)。聚类分析中使用了所有11个阶段每种代谢物的Z评分标准化值。颜色条表示每种代谢物含量的变化,从蓝色(低含量)过渡到红色(高含量)。 d 在相邻发育阶段中含量显著增加的代谢物数量及其对应的聚类信息。S1至S11分别表示花后4、6、8、10、12、14、16、18、20、22和24周的梨果实发育阶段。
为了挖掘促进果肉发育的代谢物,我们进一步分析了相邻两个发育阶段之间的差异积累代谢物(DAMs)(附加文件1:图S3a)。结果发现,与果实成熟阶段相比,早期果肉发育阶段代谢物类型的转变更加频繁(附加文件1:图S3b)。在相邻发育阶段中,共有105种代谢物显著积累,这些代谢物属于27个类别,表明它们可能在梨果肉发育中具有重要作用。
有趣的是,相邻两个阶段之间的代谢物至少涉及两个聚类(图1d)。此外,从S1到S2以及从S2到S3的代谢物转变涉及14种和11种代谢物类别,而从S9到S10以及从S10到S11的代谢物转变分别涉及7种和4种代谢物类别(表1)。这一结果表明,与果实成熟相比,果肉早期发育更依赖于代谢物的生物合成。
在果实膨大阶段(S3到S9)[47],从S4到S5以及从S6到S7的代谢物转变涉及13种和12种代谢物类别,而从S3到S4、S5到S6、S7到S8以及S8到S9的代谢物转变则分别涉及7种、5种、5种和8种代谢物类别(表1)。这些结果表明,从S4到S5以及从S6到S7的代谢物生物合成可能在果实膨大过程中起重要作用。
| Metabolite class | S1 vs S2 | S2 vs S3 | S3 vs S4 | S4 vs S5 | S5 vs S6 | S6 vs S7 | S7 vs S8 | S8 vs S9 | S9 vs S10 | S10 vs S11 |
|---|---|---|---|---|---|---|---|---|---|---|
| Alcohols and polyols | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 |
| Alkaloids | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 |
| Amino acid derivatives | 3 | 0 | 0 | 3 | 0 | 0 | 1 | 0 | 3 | 0 |
| Amino acid | 7 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 6 | 2 |
| Anthocyanins | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| Benzoic acid derivatives | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Carbohydrates | 0 | 0 | 1 | 1 | 1 | 1 | 0 | 1 | 0 | 0 |
| Catechin derivatives | 0 | 0 | 0 | 0 | 0 | 2 | 1 | 1 | 0 | 0 |
| Coumarins | 0 | 0 | 0 | 1 | 1 | 2 | 0 | 1 | 0 | 0 |
| Flavanone | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Flavone | 4 | 2 | 1 | 3 | 0 | 0 | 0 | 1 | 1 | 0 |
| Flavone C-glycosides | 3 | 1 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 |
| Flavonol | 3 | 1 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| Hydroxycinnamoyl derivatives | 3 | 3 | 0 | 1 | 0 | 3 | 0 | 0 | 0 | 0 |
| Indole derivatives | 2 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 2 |
| Lipids_Fatty acids | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 0 |
| Lipids_Glycerolipids | 2 | 0 | 0 | 0 | 1 | 4 | 0 | 0 | 1 | 0 |
| Lipids_Glycerophospholipids | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| Nucleotide and its derivates | 2 | 1 | 1 | 2 | 0 | 1 | 0 | 0 | 1 | 0 |
| Organic acids | 2 | 1 | 1 | 1 | 0 | 4 | 0 | 0 | 0 | 0 |
| Others | 1 | 1 | 2 | 2 | 0 | 1 | 0 | 0 | 0 | 0 |
| Phenolamides | 0 | 0 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 0 |
| Phytohormones | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Proanthocyanidins | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 2 | 0 |
| Quinate and its derivatives | 2 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Quinate and its derivatives | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 |
| Vitamins | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
梨果肉代谢的基因调控
为了建立基因活性与梨果肉代谢之间的关系,我们利用质谱技术分析了各阶段的蛋白质组数据。综合所有样本,共检测到5106种蛋白质,每个发育阶段平均检测到约4074种蛋白质,其中2860种蛋白质在所有阶段中均稳定存在(附加文件2:表S1)。通过比较各阶段的蛋白质丰度,共在果肉中鉴定出3469种差异表达蛋白(DEPs)(图2a)。
蛋白质组与代谢组的整合分析显示,在聚类I→VIII中,分别有1038、1012、167、843、927、287、426和162种DEPs与代谢物呈正相关(Pearson相关系数 > 0.85,假发现率 < 0.05),而分别有834、883、68、509、922、60、458和58种DEPs与代谢物呈负相关(Pearson相关系数 < −0.85,假发现率 < 0.05)(附加文件2:图S4a)。
总体而言,共有2513种DEPs与423种差异积累代谢物(DAMs)相关联(附加文件2:表S4)。其中,1581种DEPs与单一聚类中的代谢物相关联,而932种DEPs至少与两个聚类中的代谢物相关联(附加文件1:图S4b)。
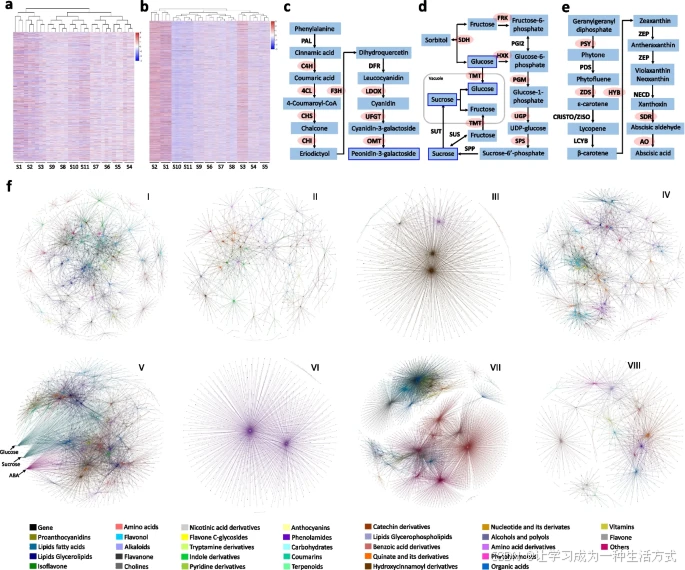
梨果肉的代谢组、蛋白质组和转录组整合分析
-
蛋白质组分析
-
a 蛋白质组聚类分析显示每个样本在任意阶段的三个可靠重复。使用11个阶段(S1至S11)每种蛋白的Z分数标准化值进行聚类分析。
-
共检测到5106种蛋白质,每阶段平均检测到4074种蛋白质,其中2860种蛋白质在所有阶段中稳定存在。
-
比较阶段间蛋白质丰度,鉴定出3469种差异表达蛋白(DEPs)。
-
-
转录组分析
-
b 转录组聚类分析显示每个样本在任意阶段的三个可靠重复,使用11个阶段中每个基因的Z分数标准化值进行聚类分析。
-
在所有样本中生成了30.6亿条原始读长(附加文件2:表S5),成功组装了33,051个基因,每阶段平均检测到28,748个基因,其中24,806个基因在所有阶段稳定存在。
-
通过阶段间基因表达水平的比较,共鉴定出22,055个差异表达基因(DEGs)。
-
-
代谢调控网络的整合
-
代谢组和转录组的整合分析表明,不同代谢物聚类(I至VIII)中的DEGs与代谢物显著正相关或负相关(Pearson相关系数>0.85或<−0.85,假发现率<0.05)。
-
综合分析鉴定出14,399个DEGs(包括2218个DEP编码基因)与439个差异积累代谢物(DAMs)相关联。
-
删除转录和翻译水平上与代谢物关联不一致的基因后,构建了全面的基因-代谢物数据库(附加文件3:表S7)。
-
-
特定代谢途径的验证
-
c 花青素生物合成途径中,芍药素-3-半乳糖苷与多个花青素生物合成基因呈正相关,如肉桂酸4-羟化酶、4-香豆酸辅酶A连接酶、查尔酮合酶、查尔酮异构酶等。
-
d 糖代谢中,蔗糖和葡萄糖与糖代谢相关基因(如山梨醇脱氢酶、果糖激酶、己糖激酶等)正相关。
-
e 脱落酸(ABA)生物合成基因(如类胡萝卜素合成酶、β-环羟化酶等)与ABA正相关。
-
f 构建了每个代谢物聚类的代谢调控网络,发现4764个基因与至少两类代谢物相关联,其中2390个基因参与了至少两个聚类。这与ABA促进可溶性糖积累的报告一致[46]。
-
-
基因-代谢物调控的染色体定位
-
将数据库中的基因定位于‘砀山酥梨’基因组的染色体上,结果显示每类代谢物的积累由位于多条染色体上的基因协同调控(附加文件1:图S6)。
-
DNA甲基化对基因表达的影响
-
全基因组甲基化测序
-
对每阶段果肉样本进行全基因组亚硫酸盐测序,生成了8.67亿条原始读长,其中97.80%用于分析全基因组DNA甲基化位点(附加文件3:表S9)。
-
在所有样本中共检测到39,016,459个胞嘧啶甲基化位点,每阶段平均27,345,777个位点被甲基化,其中18,634,952个位点在所有阶段中稳定甲基化。
-
-
甲基化动态变化
-
果肉发育过程中,全基因组和CHH区域的DNA甲基化水平逐渐增加,而CG区域的甲基化水平逐渐降低,CHG区域变化不大(附加文件3:表S9)。
-
基因体及其侧翼区域也观察到类似趋势,但基因体内的胞嘧啶甲基化水平逐渐下降(图3a)。
-
转座元件(TE)侧翼区域的CG甲基化水平逐渐降低,而TE体内及其侧翼区域的CHH甲基化水平逐渐增加(图3a)。这些结果表明,DNA甲基化的增加主要由CHH甲基化驱动,与甜橙的相关研究结果一致[26]。
-
这些发现进一步揭示了DNA甲基化在梨果肉代谢和基因调控网络中的重要作用。
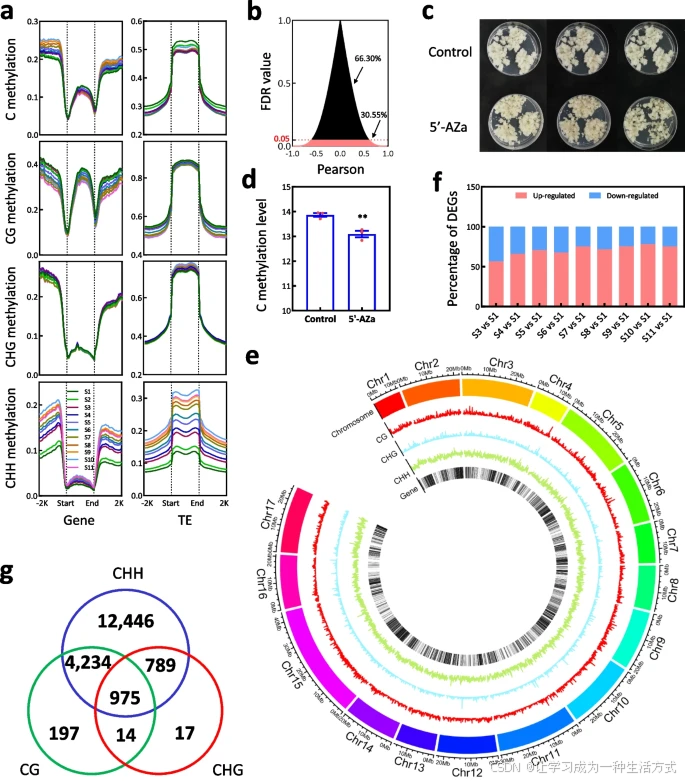
梨果肉发育过程中DNA甲基化与基因转录的关联
a. 果肉中基因与转座子(TE)在11个阶段(S1至S11)的DNA甲基化水平。 b. DNA甲基化与启动子区差异表达基因(DEGs)的相关性分析。百分比表示启动子区与DNA甲基化相关或不相关的DEGs比例。 c. 使用DNA甲基化抑制剂5-氮杂胞苷(5'-Aza)处理果肉愈伤组织,未处理愈伤组织作为对照。 d. 全基因组亚硫酸盐测序揭示5'-Aza处理和对照愈伤组织中的DNA甲基化变化。误差条基于三个重复样本计算,方差分析通过学生t检验完成,单星号表示显著性水平为P < 0.05。 e. DNA甲基化位点及所有预测基因的染色体定位,揭示了受三种甲基化(CG、CHG和CHH)修饰的基因。Chr 1至Chr 17表示梨基因组的17条染色体。 f. 图示显示S1与后续阶段相比上调和下调基因的比例。这些基因已被证实在果肉愈伤组织中受到5'-Aza处理的抑制,并在发育中的果实中受DNA甲基化修饰。 g. 韦恩图显示由CG、CHG和/或CHH甲基化修饰的DEGs数量。
DNA甲基化对基因表达的影响
为研究DNA甲基化对基因表达的影响,我们分析发现胞嘧啶甲基化存在于38,159个基因的启动子区域中,其中包括20,322个DEGs(附加文件2:图S7a)。这些DEGs中,6209个的表达水平与其启动子中的胞嘧啶甲基化平均水平呈正或负相关(假发现率FDR < 0.05;图3b,附加文件3:表S10)。这表明DNA甲基化对基因表达可能具有正负两方面的影响。
为验证这一点,用50 mM的DNA甲基化抑制剂5'-Aza处理梨果肉愈伤组织(图3c)。对5'-Aza处理和对照愈伤组织进行全基因组亚硫酸盐测序,生成了6.34亿原始读数(附加文件4:表S11)。结果显示,5'-Aza处理显著降低了胞嘧啶甲基化水平(P值=0.0071,< 0.05;图3d)。随后,RNA测序鉴定出1426个DEGs(附加文件4:表S13)。在这些基因的启动子区域检测到58,943个差异甲基化区域(DMRs),其中1328个DEGs与这些区域相关,包括705个存在于基因-代谢物数据库中的DEGs(附加文件4:表S13)。这些结果表明,这些基因的表达水平可能由果肉愈伤组织中DNA甲基化的变化决定。
发育中果肉中DEGs启动子区域的甲基化
通过分析富含胞嘧啶甲基化位点的区域,共在果肉中35,176个基因的启动子区域检测到178,499个DMRs(图3e,附加文件2:表S2)。其中92,912个位点位于18,672个DEGs的启动子区域(附加文件4:表S14),这些DEGs中有12,396个与437个DAMs相关(附加文件1:图S7b,附加文件4:表S15)。此外,647个DEGs在5'-Aza处理和对照愈伤组织中表现出差异表达(附加文件4:表S13)。将这些基因与发育中的果实中(S1与S3至S11)进行比较发现,有56.67%至78.17%的基因在S1后期表达水平较高(图3f)。这些结果表明,这些18,672个DEGs的表达可能由果肉中的DNA甲基化修饰。
值得注意的是,这些DEGs中有95.58%受到CHH DMR修饰(图3g),表明CHH DMR可能在调控果肉基因表达中比CG和CHG DMR起更重要作用。
果肉发育中DNA甲基化水平的增加与DNA去甲基化相关基因的表达减少
为揭示果肉发育过程中DNA甲基化增加的潜在机制,我们利用系统发育分析鉴定了RdDM途径相关基因(附加文件1:图S8a-d)。结果表明,PbDCL2.2未在果肉中表达(附加文件4:表S16)。整体上,PbAGO4.1、PbAGO4.2、PbAGO4.3、PbAGO6、PbDCL2.1、PbDCL3.1、PbDCL3.2、PbDCL3.3、PbDCL4、PbNRPD1、PbNRPE1、PbRDR2.1、PbRDR2.2和PbRDR2.3的表达水平在果肉发育过程中逐渐下降(附加文件1:图S8e),并且大部分与C和CHH甲基化水平呈负相关(附加文件1:图S8f)。相反,PbDCL2.3、PbDCL2.4、PbDCL2.5和PbDCL2.6的表达水平逐渐增加,尤其是PbDCL2.3与C和CHH甲基化水平呈正相关(附加文件1:图S8f)。
然而,小RNA组学分析显示,尽管4个DCL基因的表达增加,但在果肉中并未观察到24-nt siRNAs显著积累。这表明RdDM途径中的基因表达变化与果肉发育过程中DNA甲基化增加无关。
DNA甲基转移酶的表达与DNA甲基化增加的关系
植物DNA甲基化的维持由DNA甲基转移酶调控[27]。在梨基因组中,我们鉴定了12个与拟南芥同源的DNA甲基转移酶基因(附加文件1:图S10a)。PbDRM1.1、PbDRM1.2、PbDRM3、PbCMT2.1、PbCMT2.2、PbCMT2.3、PbCMT2.4、PbCMT3.1和PbCMT3.2在S1和S2阶段的表达水平高于其他阶段(附加文件1:图S10b),但与C和CHH甲基化水平呈负相关(附加文件1:图S10c)。此外,PbMET1.2在S3和S5阶段的表达高于S10和S11(附加文件1:图S10b),但与C和CHH甲基化水平无关(附加文件1:图S10c)。相反,PbMET1.1的表达水平在果肉发育过程中逐渐增加(附加文件1:图S10b),并与C和CHH甲基化水平呈正相关(附加文件1:图S10c)。
尽管如此,CG甲基化水平在果肉发育过程中逐渐降低(图4a),并与PbMET1.1的表达呈负相关(FDR = 0.002 < 0.05)。因此,DNA甲基转移酶基因的表达变化与果肉发育过程中DNA甲基化增加无关。
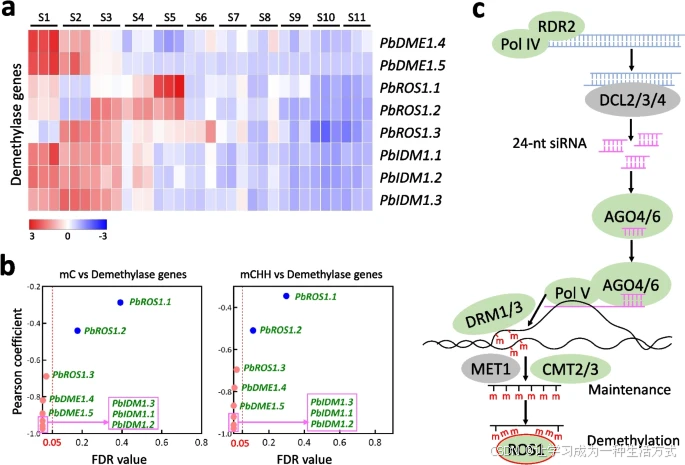
DNA去甲基化基因表达下降与梨果肉发育过程中DNA甲基化水平升高一致
-
去甲基化基因的表达模式
-
a 梨果肉在11个阶段(S1至S11)的去甲基化基因表达模式。聚类分析使用了各基因的Z分数标准化值,颜色条表示基因表达水平从蓝色(低)到红色(高)的变化。
-
b 去甲基化基因与胞嘧啶(C)甲基化(左)和CHH甲基化(右)的相关性分析。红色虚线表示假发现率为0.05的显著性阈值。
-
c 果肉中DNA甲基化的建立、维持和去除机制的示意图。
-
植物甲基化组的调控涉及DNA甲基化与去甲基化过程之间的动态平衡[27]。为了确定梨果肉中DNA去甲基化基因的表达模式,我们从梨基因组中鉴定出拟南芥ROS1基因的8个同源基因(附加文件1:图S11)。其中,PbDME1.1、PbDME1.2和PbDME1.3在果肉中未表达(附加文件4:表S16)。整体上,PbDME1.4、PbDME1.5和PbROS1.3的表达水平在果肉发育过程中逐渐下降(图4a),并与C和CHH甲基化水平呈负相关(图4b)。此外,PbROS1.1和PbROS1.2的表达水平从S5至S11逐渐降低(图4a)。这些结果表明,这些基因的表达下降与果肉发育过程中DNA甲基化水平升高一致(图4c)。
考虑到拟南芥IDM1基因通过催化组蛋白H3K18乙酰化,促进ROS1功能的作用[35],我们从梨基因组中鉴定出IDM1基因的三个同源基因(附加文件1:图S11)。PbIDM1.1、PbIDM1.2和PbIDM1.3的表达水平也在果肉发育过程中逐渐降低(图4a),并与C和CHH甲基化水平呈负相关(图4b)。因此,所有三个IDM基因表达的下降与果肉发育过程中DNA甲基化水平升高一致。综上所述,这些结果表明,梨果肉发育过程中DNA甲基化水平的升高可能与参与DNA去甲基化的基因表达下降有关。
DNA甲基化参与梨果实代谢
通过分析,鉴定出12,396个被差异甲基化区域(DMR)修饰的差异表达基因(DEGs)与437个差异积累代谢物(DAMs)相关联(附加文件4:表S15)。为了明确这些DAMs与DEGs启动子区域DNA甲基化之间的关系,相关性分析显示3987个DEGs启动子区域的DNA甲基化与所有八个聚类中的316个DAMs相关(假发现率<0.05,附加文件4:表S18)。此外,发现全基因组DNA甲基化与聚类I、II和IV的DAMs呈负相关,但与聚类V的DAMs呈正相关(附加文件4:表S19)。这些结果表明,DNA甲基化可能通过影响代谢物生成相关基因的表达,调控果肉代谢物。
进一步分析发现,大部分与ABA、蔗糖和葡萄糖的合成和代谢相关的DMR修饰DEGs已被鉴定(附加文件4:表S15)。部分DEGs启动子区域甚至全基因组的DNA甲基化与ABA、蔗糖和葡萄糖相关(附加文件4:表S18和S19)。ABA是一种重要的植物激素,通过增加可溶性糖(包括蔗糖和葡萄糖)含量以及促进乙烯释放来加速梨果实成熟[46]。因此,DNA甲基化可能通过影响ABA的生物合成参与梨果肉发育。
DNA甲基化抑制实验验证 为验证上述假设,在S9阶段对果肉表皮下施用DNA甲基化抑制剂5’-Aza(图5a)。全基因组亚硫酸盐测序显示,5’-Aza处理显著降低了胞嘧啶甲基化水平(p值=0.038<0.05,图5b)。与对照组相比,5’-Aza处理促进了果皮叶绿素积累(图5c),使果皮呈绿色(图5a)。
此外,对5’-Aza处理的果肉全基因组亚硫酸盐测序结果显示,胞嘧啶甲基化水平显著降低(p值=0.012<0.05,图5b)。与对照组相比,5’-Aza处理促进了果肉中ABA、β-胡萝卜素和叶黄素的生物合成(图5c)。这些结果表明,DNA甲基化参与类胡萝卜素代谢,并可能通过增加ABA的产生加速果实成熟。
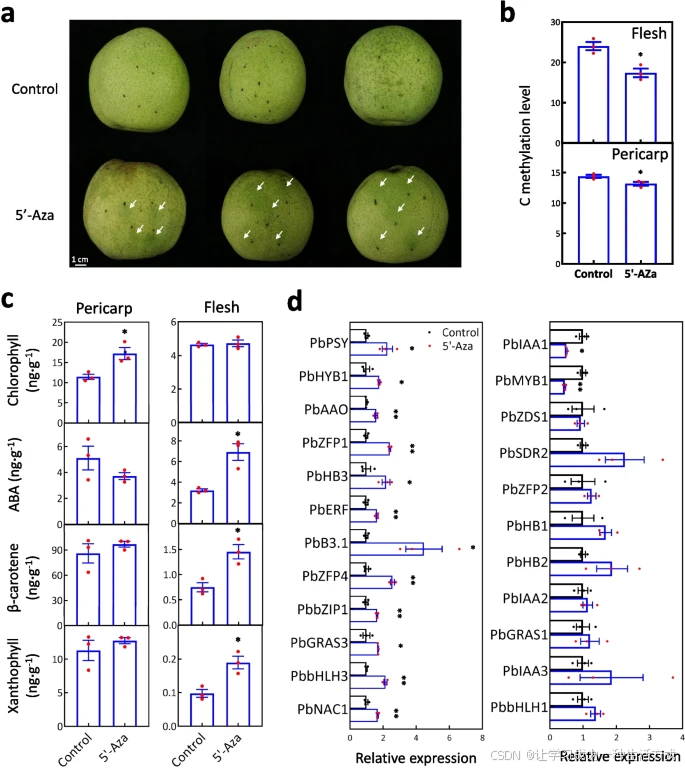
DNA甲基化在果肉代谢中的作用
-
5’-Aza处理对果实表型和DNA甲基化的影响
-
a 与H₂O对照组相比,5’-Aza处理后的梨果实表现出显著的表型变化,果皮呈深绿色(白色箭头所示)。
-
b 全基因组亚硫酸盐测序显示,5’-Aza处理后梨果实的果皮和果肉中DNA甲基化水平发生显著变化。
-
c 在果肉和果皮中测定叶绿素、ABA、β-胡萝卜素和叶黄素含量。5’-Aza处理组的这些代谢物含量较对照组显著增加。
-
d 检测了ABA生物合成基因及其正相关转录因子(TFs)的表达水平。标准误差基于三个重复计算,单星号和双星号分别表示显著性水平为P < 0.05和P < 0.01(Student’s t检验)。
-
为了阐明DNA甲基化对类胡萝卜素代谢的调控作用,在5’-Aza和H₂O处理的果肉中进行定量实时聚合酶链式反应(qRT-PCR)分析。
-
基因表达分析
-
在DMR修饰的差异表达基因(DEGs)中,10个通路基因和35个转录因子的表达水平与ABA含量呈正相关(附加文件4:表S15)。
-
其中,PbPSY、PbZDS1、PbHYB1、PbSDR2和PbAAO在S5至S11阶段的果肉中表达水平较S1至S4阶段显著升高(附加文件1:图S12)。
-
相比对照组,5’-Aza处理显著上调了PbPSY、PbHYB1和PbAAO的表达,但对PbZDS1和PbSDR2的表达无显著影响(图5d)。这些结果表明,DNA甲基化可能通过抑制PbPSY、PbHYB1和PbAAO的表达来限制果肉中ABA的产生。
-
在35个转录因子中,18个在S5至S11阶段的果肉中相对高表达(附加文件1:图S12)。相比对照组,5’-Aza处理显著上调了PbZFP1、PbHB3、PbERF、PbB3.1、PbZFP4、PbbZIP1、PbGRAS3、PbbHLH3和PbNAC1的表达,而对PbZFP2、PbHB1、PbHB2、PbIAA2等转录因子的表达无显著影响(图5d)。这些结果表明,DNA甲基化水平降低可能通过促进多个转录因子的表达,诱导PbPSY、PbHYB1和PbAAO的表达。此外,PbIAA1和PbMYB1在5’-Aza处理组中的表达水平显著下降,表明降低DNA甲基化也可能抑制某些基因的表达。
-
新转录因子在ABA生物合成中的调控作用验证
为了进一步明确测试的转录因子在ABA生物合成中的潜在作用,进行了基于ABA生物合成基因启动子的双荧光素酶实验。
-
基于表达和DNA甲基化分析,选择了12个转录因子进行测试(图5d,附加文件4:表S15)。
-
结果显示,在PbPSY、PbZDS1、PbHYB1、PbSDR2和PbAAO启动子调控下的LUC基因,均被至少三个测试转录因子显著激活(图6a)。
-
这些结果表明,这些转录因子可能激活一部分ABA生物合成相关基因。
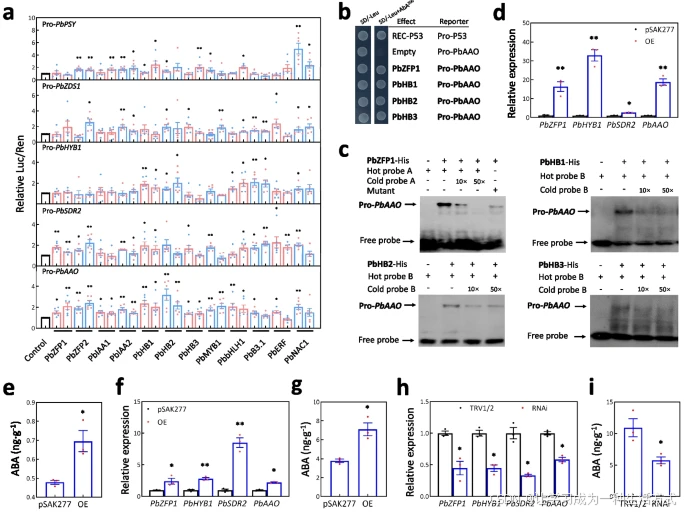
PbZFP1 正向调控果肉中 ABA 的生物合成
-
双荧光素酶实验揭示 PbZFP1 对 ABA 生物合成基因的调控作用
-
a 双荧光素酶实验显示,12个选定的转录因子(TFs)对 ABA 生物合成基因启动子的调控效应。标准误差基于至少五个重复计算,并进行了两次独立实验。
-
b 酵母单杂交实验(Y1H)显示,PbZFP1、PbHB1、PbHB2 和 PbHB3 能结合在 PbAAO 启动子的上游区域(-500 bp 至 -100 bp 之间)。
-
c 电泳迁移率变动实验(EMSA)进一步验证 PbZFP1、PbHB1、PbHB2 和 PbHB3 与 PbAAO 启动子区域的直接结合能力。其中,“+”和“–”分别表示是否存在重组的转录因子蛋白、生物素标记探针、冷探针或生物素标记突变探针。冷探针浓度为标记探针浓度的 10 倍和 50 倍。
-
AAO 是催化脱落酸醛(abscisic aldehyde)转化为 ABA 的关键酶(图2e)。
-
双荧光素酶实验显示,9个转录因子(涉及5个家族)可显著增强 PbAAO 启动子的活性(图6a)。其中,ZFP、Homeobox、MYB 和 bHLH 家族的转录因子被报道参与果实成熟[48–51]。
-
进一步实验发现,PbMYB1 和 PbbHLH1 的可能结合区域分别位于 PbAAO 启动子的上游 -1000 bp 至起始密码子、-2000 bp 至 -500 bp 之间(附加文件1:图S13a、b)。PbHB1 和 PbHB2/PbHB3 的结合区域分别位于上游 -300 bp 至 -100 bp 和 -200 bp 至 -100 bp 之间(附加文件1:图S13c)。
-
然而,由于 C2H2 型 ZFP 结合位点分布在 PbAAO 启动子的多个位置,难以明确定位 PbZFP1 和 PbZFP2 的结合区域(附加文件1:图S13b-d)。
-
酵母单杂交实验和电泳迁移率变动实验进一步确认 PbZFP1 与 PbAAO 启动子区域的直接相互作用,同时 PbHB1、PbHB2 和 PbHB3 也能直接结合(图6b、c)。这些结果表明,这些转录因子可通过与 PbAAO 启动子的物理相互作用增强其活性。
-
PbZFP1 在 ABA 生物合成中的功能验证
-
核定位的 PbZFP1 被选择用于功能验证(附加文件1:图S14)。
-
过表达实验
-
将由 35S 启动子驱动的 PbZFP1 过表达载体引入根癌农杆菌,并用于感染梨果肉愈伤组织以产生阳性转基因愈伤组织(附加文件1:图S15a)。
-
在过表达 PbZFP1 的愈伤组织中,PbZFP1、PbHYB1、PbSDR2 和 PbAAO 的表达水平以及 ABA 含量均显著高于感染空载体 pSAK277 的愈伤组织(图6d、e)。
短暂转化实验
-
将含 PbZFP1 过表达载体的农杆菌注射到 S9 阶段的‘砀山酥梨’果肉中(附加文件2:图S15b)。
-
结果显示,与感染空载体 pSAK277 的果肉相比,过表达 PbZFP1 的果肉中 PbZFP1、PbHYB1、PbSDR2 和 PbAAO 的表达水平及 ABA 含量均显著增加(图6f、g)。
病毒诱导基因沉默(VIGS)实验
-
将 PbZFP1 的 VIGS 载体引入农杆菌,并用于注射 S9 阶段的‘砀山酥梨’果肉(附加文件1:图S15c)。
-
结果显示,与感染空载体 pTRV1 和 pTRV2 的果肉相比,PbZFP1、PbHYB1、PbSDR2 和 PbAAO 的表达水平及 ABA 含量在沉默 PbZFP1 的果肉中显著下降(图6h、i)。
这些结果表明,PbZFP1 通过正向调控 ABA 生物合成相关基因的表达,介导果肉中的 ABA 生物合成。
讨论
果实发育与代谢物的积累与分解 果实发育受多种代谢物的积累和分解影响。对肉质果实(如梨和番茄)发育过程中代谢物积累的研究主要集中于糖、有机酸、黄酮和多酚等类别[6,7,8]。随着广泛靶向代谢组学方法的出现,鉴定了大量代谢物(>400种),为探索果实代谢及其调控网络奠定了基础[2,17]。例如,在番茄中,从442个熟期样本的果皮中鉴定了980种代谢物,包括362种注释代谢物,其中371种代谢物与970个SNP和535个基因相关(通过mGWAS和eQTL分析重叠确定)[2]。此外,从20个主要的番茄组织和发育阶段中检测到540种代谢物,这些代谢物与全基因组基因表达模式的相关性被用于重构番茄代谢调控网络[17]。 本研究采用了广泛靶向代谢组方法,从梨果肉11个发育阶段(从幼果到成熟)中鉴定出492种注释代谢物,与之前在桃果实中的鉴定结果相似[5]。其中,449种代谢物在不同阶段表现出显著差异积累(附加文件2:表S3)。为探究这些差异积累代谢物(DAMs)的调控网络,我们进行了蛋白质组和转录组分析,鉴定出相关基因,并构建了一个包含439个DAMs和14,399个差异表达基因(DEGs)的基因-代谢物关联数据库(附加文件3:表S7),为研究梨果肉发育中的代谢调控网络提供了重要资源。
验证已知网络与发现新网络 构建的基因-代谢物数据库既验证了已知的代谢调控网络,又发现了新的调控网络。例如,以前的研究确认了花青素生物合成由合成基因和MYB-bHLH-WD40复合体介导[52,53,54]。本研究观察到花青素生物合成基因、MYB-bHLH-WD40复合体成分与芍药素-3-半乳糖苷(一种特定花青素成分)含量之间的相似表达模式(附加文件3:表S7),进一步支持这些基因及复合体在花青素生物合成中的作用。 此外,该数据库还用作一个非偏向资源,用于识别参与ABA生物合成的新TFs。ABA通过增加可溶性糖和乙烯的含量,促进梨果实成熟[44]。已有研究表明,ERF、NAC和TCP等TF家族在ABA生成和果实成熟中发挥正向调控作用[55,56,57]。本研究鉴定出37个TF,其表达模式与ABA含量相关(附加文件3:表S7)。其中,PbZFP1、PbHB1、PbHB2和PbHB3直接结合到PbAAO启动子上以增强其活性(图6a–c)。过表达PbZFP1显著提高了果肉愈伤组织和果肉中的PbAAO表达和ABA含量(图6d–g),而沉默PbZFP1则导致PbAAO表达和ABA含量下降(图6h, i)。这些结果表明,PbZFP1作为一种新的调控因子,促进ABA生物合成并可能加速梨果实成熟。
DNA甲基化对基因表达和果实成熟的作用 DNA甲基化在基因表达调控和果实成熟中具有重要作用[24,25,26]。例如,在番茄和草莓中,DNA去甲基化水平下降通过上调成熟相关基因并下调成熟抑制基因,加速果实成熟[24,25];而在甜橙中,5’-Aza处理通过上调Cs3g20300(一种成熟诱导基因)的表达,抑制果实成熟[26]。 与番茄和草莓等一年生植物不同,梨等多年生木本植物的果实由花托膨大形成,而甜橙果实由子房发育而来。本研究中,利用DNA甲基化抑制剂5’-Aza处理愈伤组织和未成熟果肉,发现5’-Aza处理的愈伤组织中93.17%的DEGs表现出甲基化水平的显著下降(附加文件4:表S13)。在果肉中,qRT-PCR分析显示,24个基因中12个基因表达水平上升,2个基因表达水平下降(图5d)。这些结果表明,DNA甲基化在梨果肉发育过程中既可正向也可负向调控基因转录,与以往研究一致[24,26]。
DNA甲基化对果实代谢的调控作用 DNA甲基化在桃果实中调控花青素积累[58]。本研究显示,DMR修饰的DEGs占与437个DAMs相关的全部DEGs的66.30%(图3b,附加文件4:表S15),表明DNA甲基化参与梨果肉代谢。进一步测定5’-Aza处理和对照果肉中的ABA、β-胡萝卜素和叶黄素含量,发现处理组显著升高(图5c)。同时,ABA生物合成基因(如PbPSY、PbHYB1和PbAAO)以及转录调控因子(如PbZFP1和PbHB3)的表达水平也显著提高(图5d)。因此,DNA甲基化通过调控ABA途径基因的表达,参与果肉代谢。
DNA甲基化模式与功能的差异 有趣的是,梨果肉发育过程中,ABA含量和DNA甲基化水平均逐渐升高,但降低DNA甲基化却促进了ABA积累(图5c)。类似地,在0℃条件下,与16℃条件相比,桃果实中的花青素含量和DNA甲基化水平均下降,但降低DNA甲基化促进了花青素积累[58]。这些结果与番茄和甜橙的研究结果存在差异,表明仅通过DNA甲基化与果实表型的相关性分析,难以全面理解其作用模式。基于本研究结果,DNA甲基化在木本植物(梨和甜橙)果实发育中呈上升趋势,而在一年生植物(草莓)中则下降。是否这一模式适用于其他木本和草本植物尚需进一步证据验证。
果实发育的分子网络 本研究通过对梨果肉11个阶段的代谢组、转录组、蛋白质组和DNA甲基化组的综合分析,构建了果肉发育过程中的代谢调控网络。通过分析相邻阶段的DAMs和DEGs,发现相邻阶段DAMs相关的基因占相邻阶段DEGs的比例从20.77%(S11对S10)到74.32%(S5对S4)不等(图7a)。这些相关基因中,大部分(>87%)受DMR修饰(图7a,附加文件4:表S20)。这种网络通过以下方式驱动果实成熟(图7b):
-
在DNA甲基转移酶和去甲基酶的动态调控下,靶基因启动子区域发生DNA甲基化,从而影响基因转录;
-
TF受DNA甲基化调控并转录成蛋白,结合到催化酶编码基因的启动子;
-
催化酶基因在DNA甲基化和TF蛋白的调控下转录;
-
催化酶将一种代谢物转化为另一种,驱动果实从一个发育阶段过渡到另一个阶段,直至成熟。
梨果肉发育过程中,大多数与DNA甲基化和去甲基化相关的基因下调,导致胞嘧啶甲基化水平整体上升。

梨果肉发育中代谢物、基因和DNA甲基化的总结
-
代谢物和基因的数量分析
-
a 在梨果肉发育的相邻阶段之间计算代谢物和基因的数量。差异积累代谢物(DAM)从代谢组数据中计算,差异表达基因(DEG)从转录组数据中计算。
-
与相邻阶段DAM相关的基因从基因-代谢物数据库中提取,被称为“相关基因”。DEGs和相关基因的交集被定义为“相关DEGs”,而由DNA甲基化修饰的相关DEGs被称为“DMR修饰相关DEGs”。
-
S1至S11分别代表梨果肉发育的不同阶段。
-
-
分子机制模型
-
b 构建了一个模型展示发育诱导的DNA甲基化如何影响梨果肉发育的分子途径:
-
在果肉发育过程中,转录因子(TFs)诱导催化基因的表达,导致催化酶积累增加,这些酶将一种代谢物(代谢物A)转化为另一种代谢物(代谢物B),从而促进果肉的发育过程。
-
同时,这一过程抑制了去甲基化酶、RNA引导的DNA甲基化(RdDM)途径及甲基转移酶基因的表达,导致DNA甲基化水平升高。
-
升高的DNA甲基化影响了转录因子和下游催化基因的转录,进而调控梨果肉的发育。
-
-
结论
综上所述,建立的基因-代谢物数据库为探索代谢调控网络提供了支持,并为提高梨果实质量奠定了基础。在梨果肉发育过程中,DNA甲基化水平的升高通过调控ABA生物合成分子网络中的基因,抑制了发育诱导的ABA积累。这表明DNA甲基化通过介导果肉代谢参与了梨果肉的发育过程,并可能延缓果实的成熟。
方法
植物材料
-
使用砀山酥梨(Pyrus bretschneideri)栽培品种,植株种植于南京农业大学江浦果园(中国南京)。
-
从开花后4周起,每隔2周采摘果实,直至果实成熟,共11个不同发育阶段。
-
样本采自一棵12年生梨树,每次采集12–16个果实,并分为三组,每组至少包含4个果实。
-
每组果实去皮后,将果肉切成小块混合。混合样本立即用液氮冷冻并储存于−80℃以供后续使用。
代谢物测定
-
样本处理
-
称取20 ± 0.2 g储存样本,进行冷冻干燥后称重并研磨成细粉。
-
称取0.1 g干粉,溶解于1.2 mL 70%甲醇中,涡旋混合30秒,每30分钟一次,重复6次。
-
将混悬液于4℃冰箱静置过夜后,以12,000 rpm离心10分钟。
-
过滤所得上清液(孔径0.22 μm),用于测定广泛靶向的代谢物。
-
-
检测方法
-
使用液相色谱-电喷雾电离-串联质谱系统,通过多反应监测(MRM)方法对代谢物进行相对定量[59, 60]。
-
信号强度以内标(利多卡因,0.1 mg l⁻¹)归一化。广泛靶向代谢物通过标准品验证。
-
每个样本进行三次生物学重复以保证结果可靠性。
-
-
类胡萝卜素、ABA 和叶绿素测定
-
提取和定量方法参考橙子[61]、草莓[62]和梨[63]的研究。
-
类胡萝卜素和ABA通过高效液相色谱(HPLC)测定(Waters Corp., USA)。
-
ABA标准品购自Sigma公司,叶黄素和β-胡萝卜素标准品购自远野生物科技公司(上海,中国)。
-
叶绿素含量通过多功能读板仪(SpectraMax iD5, MD, USA)测定。
-
每个样本均进行三次生物学重复。
-
蛋白质提取与测序
-
样本处理
-
果肉样本在液氮中研磨成粉,每个样本三次生物学重复。
-
每个重复取150 mg冷冻干燥粉末,使用裂解液(8 M尿素,1% SDS,100 mM Tris-HCl,pH 8.0)及蛋白酶抑制剂混合物(Sigma, USA)裂解。
-
裂解液在冰上孵育20分钟后,与4倍体积的10 mM二硫苏糖醇(DTT)冷丙酮混合,−20℃静置过夜提取蛋白。
-
蛋白溶解于缓冲液(8 M尿素,100 mM Tris-HCl,pH 8.0)。
-
-
蛋白消化与纯化
-
取0.1 mg蛋白,经过DTT还原和碘乙酰胺烷基化处理后,以胰蛋白酶(Trypsin Gold, Promega, USA)在37℃孵育16小时。
-
消化后的肽段通过C18柱脱盐后,真空离心干燥。
-
-
定量测定
-
使用液相色谱-串联质谱(LC-MS/MS)系统,配备C18柱(150 µm内径,360 µm外径 × 15 cm,1.9 µm C18填料)。
-
依照先前研究[64],结合数据依赖采集(DDA)和数据非依赖采集(DIA)协议,稍作修改。
-
DDA实验设置:母离子质量范围为350–1500 m/z,离子注入时间45 ms,隔离窗口1.6 m/z,标准化碰撞能量27%。
-
DIA实验设置:全质谱分辨率为60,000,离子注入时间50 ms,质量范围为350–1200 m/z。
-
-
数据处理
-
使用Proteome Discoverer 2.2(Thermo)对DDA数据进行梨蛋白数据库比对。
-
DIA数据分析使用Skyline软件[65],通过色谱图提取前体离子和碎片离子,筛选符合标准的峰值。
-
错误率和特征分数由mProphet算法计算[66],定量分析通过MSstats程序完成[67]。
-
-
结果整理
-
将所有识别蛋白的表达模式记录于附加文件4:表S21中。
-
mRNA测序
-
使用天根公司(北京,中国)的多糖和多酚丰富植物RNA提取试剂盒提取样品总RNA。每个样本进行三次生物学重复。
-
评估RNA样品的纯度、浓度和完整性,仅RIN值大于7的样本用于文库构建和测序。
-
mRNA测序文库的构建:
-
使用Epicentre Ribo-zero™ rRNA移除试剂盒去除核糖体RNA。
-
剩余RNA使用NEBNext® Ultra™ Directional RNA Library Prep Kit(NEB,美国)处理,构建测序文库。
-
文库通过Illumina Hiseq 4000平台进行150 bp双端测序。
-
-
高质量读段(清洁读段)使用Bowtie 2 v2.2.8和HISAT2 v2.0.4 [68]映射到砀山酥梨参考基因组V1.0(Pear Genome Project),允许3个错配。
-
每个样本的映射读段使用StringTie v1.3.1 [69]进行基于参考的组装,转录本表达水平通过Cufflinks v2.1.1 [70]计算,并以每千碱基转录本每百万映射片段的读段数(RPKM)表示。
代谢物、mRNA和蛋白质的关联分析
-
对代谢物、mRNA和蛋白质在相邻阶段之间进行差异分析,筛选标准为绝对倍数变化≥2且假发现率(FDR)< 0.05。
-
DAM通过代谢物信号强度评估,DEP基于多肽的定量浓度鉴定,DEG使用edgeR生物导包[71]分析,基于映射到参考基因组的归一化读段计数。
-
使用Python脚本(https://github.com/Peims/calculate-the-pearsoner)进行相关性分析,评估代谢物与mRNA、蛋白质和DNA甲基化之间的关系。所有测试样本中使用三次重复的平均值。
-
显著相关性的阈值设置为Pearson系数> 0.85或< -0.85,FDR< 0.05。
全基因组亚硫酸氢盐测序
-
使用CTAB法提取基因组DNA,使用Nanophotometer®分光光度计(IMPLEN,美国)和Qubit® DNA Assay Kit(Life Technologies,美国)测定纯度和浓度。
-
DNA片段化为200–300 bp,并进行末端修复及加A尾处理。
-
使用EZ DNA Methylation-GoldTM Kit(Zymo Research,美国)进行两次亚硫酸氢盐处理。
-
处理后的单链DNA使用KAPA HiFi HotStart Uracil + ReadyMix(Zymo Research)扩增,并通过Illumina Hiseq 4000(美国)平台进行150 bp双端测序。
-
移除低质量读段后,剩余读段使用Bismark v0.16.3 [72]映射到砀山酥梨参考基因组,不允许错配。
-
鉴定甲基化位点并计算其水平,DMR使用DSS软件[74]鉴定。
小RNA测序
-
使用NEBNext® Multiplex Small RNA Library Prep Set(NEB)处理总RNA构建小RNA文库。
-
为每个样本分配索引码,进行两次生物学重复。
-
索引编码样本生成簇后,文库通过Illumina Hiseq 2500平台进行50 bp单端测序。
-
移除低质量读段后,筛选长度在18–30 bp之间的高质量读段,使用Bowtie [75]映射到参考基因组,不允许错配。
-
使用ShortStack [76]定义24-nt siRNA,并通过将映射读段归一化到总清洁读段计算24-nt siRNA的丰度。
蛋白-DNA相互作用
-
双荧光素酶实验(Dual-luciferase assay):
-
将每个转录因子的全长编码序列插入pSAK277载体生成过表达载体。
-
将约2000 bp启动子序列插入pGreenII 0800-LUC载体。
-
构建物转化入农杆菌GV3101菌株后,根据已发表的协议[3]测量萤火虫荧光素酶(LUC)和海藻荧光素酶(REN)活性。
-
-
电泳迁移率变动实验(EMSA):
-
将转录因子的全长编码序列插入带有His标签的pCold-TF表达载体,生成重组蛋白。
-
生物素标记探针由生工生物(上海,中国)合成,实验参照已有方法[77]进行。
-
-
酵母单杂交实验(Y1H assay):
-
将转录因子的全长编码序列插入pGADT7载体。
-
从“砀山酥梨”基因组DNA中扩增PbAAO启动子片段并插入pAbAi载体。
-
使用Matchmaker Gold酵母单杂交文库筛选系统(Clontech,美国)进行实验。
-
-
引物序列列于附加文件4:表S22。
DNA甲基化抑制剂处理
-
5’-Aza处理
-
对砀山酥梨(cv. Dangshansuli)果实的S9阶段样本和Clapp Favorite(Pyrus communis)果肉愈伤组织进行5’-Aza处理。
-
在果实中,将大约200 μL的40 mM 5’-Aza溶液(Sigma公司,溶于双蒸水)注入果肉及果皮。处理后10天拍摄样品照片。
-
在愈伤组织中,用50 mM 5’-Aza处理幼嫩愈伤组织,处理15天后拍摄样品照片。
-
-
DNA甲基化检测
-
从5’-Aza处理的果实和愈伤组织中提取基因组DNA,并使用EZ DNA Methylation-Gold™ Kit(Zymo Research)进行亚硫酸氢盐处理。
-
通过全基因组亚硫酸氢盐测序检测DNA甲基化水平。
-
-
基因表达和类胡萝卜素代谢分析
-
从未经处理和5’-Aza处理的愈伤组织中提取总RNA,按mRNA测序相同的方法进行转录组测序和分析。
-
转录组读段映射到Bartlett DH(Pyrus communis)参考基因组(GDR)。
-
测定5’-Aza处理果肉及果皮中的ABA、类胡萝卜素和叶绿素含量,并使用Microsoft Office Excel 2016计算显著性差异。
-
系统发育分析
-
通过在Phytozome数据库(https://phytozome-next.jgi.doe.gov/)中对草莓和橙子的基因组进行BLAST搜索,鉴定拟南芥中DNA甲基转移酶、DNA去甲基化酶及RdDM通路基因的梨同源基因。
-
使用MEGA 6软件,基于鉴定基因的氨基酸序列构建邻接法(Neighbor-Joining)系统发育树,采用1000次重复计算自展值。
梨果实中的转化
-
过表达研究
-
将携带PbZFP1过表达载体或空载体pSAK277的农杆菌感染Clapp Favorite愈伤组织,操作参照先前研究[78]。
-
在果实中,将农杆菌注射到S9阶段的果肉中,操作参照文献[77]。
-
愈伤组织于继代培养10天后采集,果肉和果皮在注射后15天采集。
-
-
病毒诱导的基因沉默(VIGS)实验
-
将特定片段(1260–1619 bp)扩增后插入pTRV2载体。
-
构建的载体与pTRV1一起转化入农杆菌EHA105,注射至S9阶段梨果实中。
-
共注射携带pTRV1和pTRV2的农杆菌作为阴性对照。果皮和果肉在注射后20天采集。
-
使用LightCycler 480® II/96热循环仪(Roche Diagostics)进行qRT-PCR分析,所有实验包括三次生物学重复。
-
ABA含量在瞬时过表达和沉默的果实样本中测定。
-
数据和材料的可用性
-
测序数据
-
11个阶段果肉的转录组、DNA甲基组及小RNA组测序数据分别存储于NCBI BioProject,访问号为PRJNA1074939[79]、PRJNA838771[80]和PRJNA838797[81]。
-
蛋白质组和代谢组数据存储于ProteomeXchange Consortium(ProteomeCentral Datasets),通过iProX存储库访问,数据集标识符为PXD049025[84]。
-
-
5’-Aza处理样本数据
-
处理和对照愈伤组织的转录组和DNA甲基组数据分别存储于NCBI BioProject,访问号为PRJNA1007363[85]和PRJNA1015450[86]。
-
处理和对照果实的DNA甲基组数据存储于NCBI BioProject,访问号为PRJNA1007412[87]和PRJNA1007414[88]。
-
-
参考基因组和数据库
-
砀山酥梨参考基因组由Pear Genome Project提供,Bartlett DH参考基因组由Genome Database for Rosaceae提供。
-
基因-代谢物数据库存储于Zenodo存储库(DOI: https://doi.org/10.5281/zenodo.10674225[91])。
-
所有分析数据可在论文和其补充文件中获取,未使用其他未提及的脚本或软件。
-


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









