










Genome sequence of the agarwood tree Aquilaria sinensis (Lour.) Spreng: the first chromosome-level draft genome in the Thymelaeceae family
沉香树(Aquilaria sinensis (Lour.) Spreng)的基因组序列:瑞香科家族中的首个染色体级草图基因组

摘要
背景 白木香(Aquilaria sinensis (Lour.) Spreng)是中国重要的沉香生产植物之一。从受损的白木香树中收集的沉香树脂自古以来一直在亚洲用于芳香或药用目的。然而,由于缺乏准确和高质量的遗传信息,沉香形成的机制仍然理解不充分。
研究发现 我们使用整合策略,结合了Nanopore、Illumina和Hi-C测序技术,报道了白木香的基因组结构。最终组装的基因组大小约为726.5 Mb,具有较高的连续性,contig N50为1.1 Mb。我们结合Hi-C数据与基因组组装,生成了染色体级别的scaffold。与8条染色体对应的8个超scaffold被组装为716.6 Mb的最终大小,使用1,862个contig,scaffold N50为88.78 Mb。BUSCO评估显示基因组完整性达到了95.27%。重复序列占59.13%,并在基因组中注释了29,203个蛋白编码基因。通过使用单拷贝直系同源基因进行的系统发育分析,我们发现白木香与锦葵目中的陆地棉(Gossypium hirsutum)和可可树(Theobroma cacao)关系密切,并且白木香在约53.18至84.37百万年前从它们的共同祖先分化出来。
结论 我们在此呈现了首个白木香的染色体级基因组组装和基因注释。本研究应为进一步研究沉香形成机制、基于基因组的改良及白木香物种的保护生物学提供宝贵的遗传资源。
背景
沉香是来自Aquilaria或Gyrinops属树木的香树脂填充的心材,其高质量制品在国际市场上的价格比黄金还要昂贵【1, 2】。沉香作为珍贵的熏香,长期用于佛教、伊斯兰教和印度教的宗教仪式中,同时也作为传统药物应用于中国传统疗法和印度的阿育吠陀【3】。现代药理学和化学研究表明,倍半萜和苯乙基色酮衍生物是沉香中的主要化合物,许多此类化合物被研究具有潜在的药理活性,包括神经保护、镇静、乙酰胆碱酯酶抑制、抗氧化、抗菌和抗炎等作用【4-7】。然而,健康的Aquilaria树木几乎不生成沉香,除非它们受到各种形式的损伤或微生物侵袭。在野外,沉香的形成通常与自然因素相关,例如风或闪电损伤,或昆虫和真菌的啃噬【8, 9】。由于沉香在医学和经济上的潜力,亚洲传统的沉香生产方法包括砍伐、钉钉子、钻孔、烧树干或修剪部分树干【10】。这导致了野生Aquilaria植物的过度开发,许多物种目前正逐渐减少或濒临灭绝【11】。
Aquilaria sinensis一直被收割和培育用于生产沉香,在中国早在公元七世纪就已用于传统中医【11】。A. sinensis的形态特征和沉香如图1所示。作为中国最大的沉香生产地,过去十年中Aquilaria sinensis的种群数量急剧下降,野生种群受到了威胁【11, 12】。由于沉香耗时的制备过程和植物资源的耗竭,沉香的供应受到限制。尽管通过转录组测序已描述了与沉香形成过程中萜烯合成或应激反应相关基因的表达【2, 13, 14】,但由于缺乏准确的基因组信息和遗传资源,沉香形成的分子机制仍然不清楚。最近发现,2-(2-苯乙基)色酮及其衍生物是Aquilaria sinensis中沉香形成的关键标志物,并已阐明了其假设的生物合成途径【8】。随着Aquilaria sinensis在野外种群的减少和沉香市场需求的增加,探索基因组背景以研究沉香形成机制并加速基因组辅助育种系统的改良显得尤为重要。

Aquilaria sinensis的形态特征。(a) 成熟树木;(b) 花;(c) 果实;(d) 种子;(e) 裂开的种子;(f) 沉香的生成;(g) 沉香。图b–e是在暗场条件下使用立体荧光显微镜(奥林巴斯SZX16,匹兹堡,PA)拍摄的。所有照片均由王俊博士拍摄,并由丁旭坡博士处理。
在此研究中,我们通过Illumina短读序列、Oxford Nanopore Technologies (ONT)长读序列和Hi-C数据的混合方法对Aquilaria sinensis(NCBI:txid210372)基因组进行了测序和组装。我们揭示了Aquilaria sinensis的基因组特征,包括重复序列、基因注释和进化。这个参考基因组将为阐明沉香的代谢形成提供基础的遗传信息,并促进对沉香树的遗传研究。
数据描述
基因组DNA提取与基因组大小估算
从中国海口市城西区(经度110 19.245 E,纬度19 59.757 N)采集了一株Aquilaria sinensis(Lour.)Spreng栽培品种的植株样本。采集后,将健康的新鲜叶片用液氮快速冷冻,然后在实验室中于−80°C下保存,直到进行DNA提取。使用改良的CTAB方法【15】从这些叶片中提取高分子量植物基因组DNA。通过在0.75%的琼脂糖凝胶上进行电泳以及使用NanoDrop D-1000分光光度计(NanoDrop Technologies, Wilmington, DE)检查提取DNA的质量和数量,然后使用Qubit技术(Life Technologies, Carlsbad, CA)对DNA进行精确定量。随后,使用标准操作流程在Illumina Hiseq2500平台(Illumina HiSeq 2500系统,RRID:SCR_016383)上构建了插入片段长度为270 bp的150 bp双末端(PE)文库,生成了49.84 Gb的原始数据。使用以下公式估算了Aquilaria sinensis的基因组大小: 基因组大小 = [Num (总k-mer数) − Num (错误k-mer数)] / k-mer的平均深度【16, 17】。 最终估算的Aquilaria sinensis基因组大小为773.3 Mb,总19-mer数约为3.71 × 10¹⁰,19-mer的峰值深度为48(补充图S1)。A. sinensis基因组的GC含量为39.23%,属于中等GC水平(补充图S2)。同时,A. sinensis基因组的杂合率为0.6%,重复序列含量为53.12%【18】。
基因组测序和使用Nanopore长读序列的组装
我们使用Oxford Nanopore SQK-LSK 108试剂盒和GridION协议(Oxford Nanopore Technologies,英国牛津)【19】制备了一个Nanopore 1D文库。首先,用NEBNext FFPE修复混合物(New England Biolabs [NEB])和NEBNext Ultra II末端修复/dA尾加成模块(NEB)对基因组DNA进行修复和末端准备。接着,使用AMPure XP磁珠(Beckman Coulter)纯化DNA,并使用由ONT提供的测序接头与高浓度T4 DNA连接酶(NEB)连接。通过使用稀释缓冲液(ONT)和洗涤缓冲液(ONT)进行AMPure XP磁珠纯化后,文库与测序缓冲液(ONT)和文库加载磁珠(ONT)混合,并加载到GridION X5平台的16个流动槽(R9.4)上(GridION,RRID:SCR_017986)【20】,生成了71.3 Gb的原始DNA读段(相当于基因组组装的约100倍覆盖率)。在去除接头序列后,我们获得了480万条Nanopore长读序列(共67.7 Gb),N50读长为21.29 kb,最长读长为935.06 kb(补充表S1)。
通过wtdbg(wtdbg,RRID:SCR_017225)1.3版【21】对从Nanopore获得的干净长读段进行初始组装,参数设置如下:wtdbg -t 60 -i Passed.fastq -o Sample -H -k 17 -S 1.01 -e 4。接着,使用Pilon 1.22版(Pilon,RRID:SCR_014731)【22】和BWA(BWA,RRID:SCR_010910)【23】进行三次迭代的抛光,均使用默认参数。Pilon程序还使用默认参数运行,以填补缺口、修复碱基(包括单核苷酸多态性和插入缺失)并修正局部组装错误。最终,99.26%的Illumina短读段能够与组装的基因组对齐(补充表S2)。初步草图基因组组装为720 Mb,contig N50长度为1.1 Mb,最长的contig长度为11.9 Mb(补充表S3)。Aquilaria sinensis基因组的contig N50远高于其他已发表的药用植物基因组组装结果(补充表S4)。
Hi-C文库构建和染色体级别组装
Hi-C技术源自染色体构象捕获技术,它通过结合基于邻近性的连接与大规模并行测序,探测整个基因组的三维结构【24】。Hi-C互作矩阵已被广泛用于组装校正,以生成染色体级的scaffold。在本研究中,使用标准方法从Aquilaria sinensis的新鲜叶片样本中提取用于Hi-C文库的基因组DNA。裂解细胞后的交联DNA用甲醛固定后,用DpnII酶切割。粘性末端被生物素标记并通过邻近连接形成嵌合连接,然后被物理剪切成300–500 bp大小的片段。在PCR扩增后,将代表原始交联和长距离物理相互作用的嵌合片段处理成PE测序文库。PCR循环程序如下:95°C 5分钟;循环18次;4°C 30秒,45°C 1秒,70°C 20秒,98°C 30秒;最后保持在4°C。根据Hi-C协议,PCR产物被纯化,然后纯化的DNA被剪切、末端修复、加上腺嘌呤尾巴并连接通用接头,并按制造商推荐的方法对样品进行索引【25】。
全基因组Hi-C文库在Illumina Hiseq 2500平台上使用150 bp PE测序进行测序。使用Fastp(版本0.12.6)和HiC-Pro(HiC-Pro,RRID:SCR_017643)【26, 27】过滤接头和低质量读段后,生成了714.27百万条干净的PE读段(约103.07 Gb,大约为组装基因组的142倍覆盖率)。通过使用bowtie2(bowtie2,RRID:SCR_005476)【28】将Hi-C数据与基于Nanopore的组装进行比对,我们发现了93.49百万条唯一比对的PE读段和62.89百万条有效的相互作用对,分别占干净数据的26.18%和17.61%(补充表S5)。我们使用BWA和Lachesis(Lachesis,RRID:SCR_017644)软件比对PE读段,并保留比对到距每个限制酶切位点500 bp远的读段【29】。根据聚类、排序和定向组装contig的方法,这些序列被分为8个染色体簇,并使用Lachesis软件通过调整参数进行scaffold构建(补充表S6,图2)。最终,使用R(版本3.5.3)【30, 31】生成了最终组装的Hi-C互作热图。
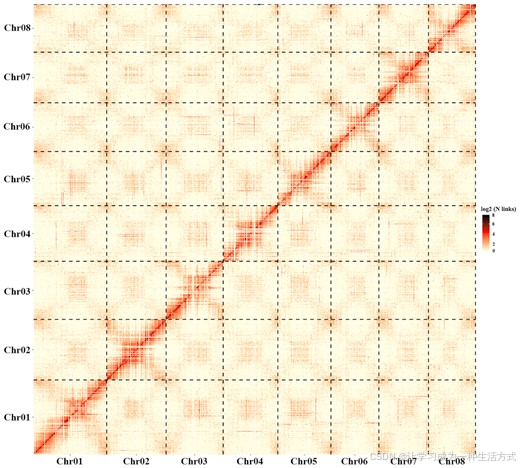
共使用了1,862个contig进行Hi-C数据辅助的scaffold构建,最终生成了805个scaffold。通过Hi-C辅助生成的染色体级scaffold的总大小为716.6 Mb,占草图基因组的99.85%,显示了很高的连续性,contig N50为1.1 Mb,scaffold N50为88.78 Mb。Aquilaria sinensis的最终草图基因组组装大小为726.5 Mb(补充表S3)。基于Hi-C组装,contig(大于100 kb)锚定到拟染色体的比例高达98.63%(表1)。A. sinensis基因组的scaffold N50也优于其他已发表的药用植物基因组组装(补充表S4)。
| Statistic | Contig length (bp) | Contig No. | Scaffold length (bp) | Scaffold No. |
|---|---|---|---|---|
| N50 | 1,058,652 | 164 | 88,784,932 | 4 |
| N60 | 726,407 | 246 | 86,380,100 | 5 |
| N70 | 495,861 | 366 | 84,956,755 | 6 |
| Longest | 11,913,571 | 1 | 109,870,270 | 1 |
| Total | 720,187,708 | 2,015 | 726,587,161 | 9 |
| Length ≥1 kb | 720,187,482 | 2,013 | 726,587,161 | 9 |
| Length ≥2 kb | 720,179,880 | 2,008 | 726,587,161 | 9 |
| Length ≥5 kb | 720,112,854 | 1,991 | 726,587,161 | 9 |
RNA制备与测序
为了基因组组装和注释,进行了Iso-seq测序。用于RNA提取的根、茎和叶混合样品来自与进行Oxford Nanopore DNA测序相同的植株,并立即用液氮快速冷冻。使用Qiagen RNA提取试剂盒(Qiagen, Hilden, DE)从冷冻组织中提取总RNA,随后使用SMRTbellTM模板制备试剂盒1.0(Pacific Biosciences, Menlo Park, CA, USA)进行测序文库的制备,之前通过SMARTerTM PCR cDNA合成试剂盒进行RNA逆转录,并使用KAPA HiFi PCR试剂盒(Kapa Biosystems, Boston, Massachusetts, USA)扩增cDNA。随后,使用PacBio Sequel系统(PacBio Sequel System, RRID:SCR_017989)进行了全长转录组测序。在通过SMRTLING 5.1过滤原始数据后,Iso-seq共获得了18,411,342条子读段,并得到了136,050条共识序列,其中94.71%(128,854条)能够与Aquilaria sinensis的最终基因组对齐(补充表S7)。
基因组质量评估
为了评估组装的完整性,我们使用了BUSCO第3版(BUSCO, RRID:SCR_015008) (BUSCO, Embryophyta odb 10)【32, 33】对最终组装的基因组序列进行了评估。总体上,在我们的基因组组装中,鉴定出了95.27%的1,375个预期的植物基因,作为完整和部分的BUSCO轮廓。在鉴定出的1,310个完整的预期植物基因中,1,202个被鉴定为单拷贝基因,108个为重复基因(补充表S8)。
Aquilaria sinensis基因组组装中的重复序列
通过基于同源性注释和de novo方法识别了转座元件(TEs)和串联重复序列。使用软件包RepeatModeler版本1.04(RepeatModeler, RRID:SCR_015027)【34】de novo识别并分类了重复元素的共识序列。使用RepeatMask版本3.2.9(RepeatMasker, RRID:SCR_012954)【34】,RepeatProteinMasker【35】,和TRF【36】在各个基因组中发现并识别了重复序列。此外,还使用MISA(MISA, RRID:SCR_010765)【37】对Aquilaria sinensis基因组中的简单重复序列(SSRs)进行了分类。结果显示,与Repbase【38】预测的重复序列相比,de novo预测的重复序列活跃时间较近(补充图S3)。在Aquilaria sinensis基因组组装中,识别出的重复序列占59.13%,其总长度为425.87 Mb(补充表S9)。特别是,详细结果表明长末端重复序列(LTRs)是最丰富的重复类型,而两类非LTR逆转座子,短散在核元件(SINEs)和长散在核元件(LINEs)【39】,在最终组装中所占比例最低。此外,13.12%的重复序列无法分类(表2)。从草图组装中的675个scaffold中共鉴定出367,251个SSR。在我们的组装中,单核苷酸(64.71%)、二核苷酸(18.19%)和三核苷酸(12.46%)构成了SSR中的近96%(补充表S10)。
| Type | Repbase TEs | Mips-REdat TEs | TE proteins | RepeatModeler | Combined TEs | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Length (Mb) | % in genome | Length (Mb) | % in genome | Length (Mb) | % in genome | Length (Mb) | % in genome | Length (Mb) | % in genome | |
| DNA | 13,223,408 | 1.84 | 1,392,136 | 0.19 | 10,456,270 | 1.45 | 28,698 131 | 3.98 | 38,895,471 | 5.4 |
| LINE | 2,916,904 | 0.41 | 253,492 | 0.04 | 7,680,548 | 1.07 | 6,394,899 | 0.89 | 12,239,695 | 1.7 |
| LTR | 73,748,923 | 10.24 | 22,973,865 | 3.19 | 75,336,839 | 10.46 | 138,348,032 | 19.21 | 192,609,862 | 26.74 |
| SINE | 2,232 | 0 | 1,145 | 0 | 0 | 0 | 0 | 0 | 4,539 | 0 |
| Other | 6,189,190 | 0.86 | 380,555 | 0.05 | 1,369,337 | 0.19 | 0 | 0 | 87,659,087 | 12.17 |
| Unknown | 35,443 | 0 | 0 | 0 | 0 | 0 | 124,331,790 | 17.26 | 94,460,416 | 13.12 |
| Total | 96,116,100 | 13.35 | 25,001,193 | 3.47 | 94,842,994 | 13.17 | 296,679,047 | 41.19 | 425,869,070 | 59.13 |
基因预测与注释
基因预测使用了三种策略。使用Augustus版本3.2.3(Augustus,RRID:SCR_008417)【40】、GlimmHmm【41】和GeneID(GeneID,RRID:SCR_002473)【42】进行ab initio基因预测,基于Corchorus olitorius(COLO4_1.0)【43】、Durio zibethinus(Duzib1.0)【44】、Gossypium hirsutum(ASM98774v1)【45】、Herrania umbratica(ASM216827v2)【46】、Theobroma cacao(Cirollo_cocoa_geneoe_v2)【47】和Arabidopsis thaliana(TAIR10)【48】的编码序列(CDS)进行模型训练。使用GeneWise(GeneWise,RRID:SCR_015054)【49】和GeMoMa【50】进行同源预测。基于表达序列标签和cDNA序列,使用PASA(PASA,RRID:SCR_014656)【51】和Tophat(TopHat,RRID:SCR_013035)【52】进行基因结构预测。最终,通过EVM【51】整合这三种策略的结果并使用Transposon PSI(Transposon,RRID:SCR_001159)【53】过滤转座元件,获得了总的基因预测结果。混合组织的RNA测序数据分别使用MatchAnnot【54】与参考基因组的注释进行了比对。
最终注释包含29,203个基因模型,转录本的平均长度为3,177.62 bp,编码区(CDS)的平均长度为1,114.16 bp,每个基因包含5.02个外显子,平均长度为222.09 bp。还计算了Aquilaria sinensis与6种相关植物的基因比较信息,包括CDS和基因长度、外显子和内含子长度及其数量(补充图S4)。通过对肽序列进行Blastall【55】和KAAS【56】搜索,推断了基因的可能功能,对比了Swiss-Prot(Swiss-Prot,RRID:SCR_002380)【57】、NR【58】、TrEMBL(TrEMBL,RRID:SCR_002380)【57】、KEGG数据库(KEGG,RRID:SCR_012773)【59】、COG数据库(COG,RRID:SCR_007273)【60】和基因本体(GO)数据库(GO,RRID:SCR_002811)【61】。蛋白质保守模型和基序预测使用InterProScan版本5.2(InterProScan,RRID:SCR_005829)【62】进行。在这29,203个编码蛋白的基因中,82.64%具有功能注释。数据库的搜索结果如下:Swiss-Prot(19,586 [67.07%])、NR(24,097 [82.52%])、TrEMBL(23,455 [80.32%])、KEGG(8,494 [29.09%])、COG(13,592 [46.54%])、GO(14,019 [48.00%])和InterProScan(20,031 [68.59%])(补充表S12)。此外,我们还通过Rfam非编码RNA数据库(Rfam,RRID:SCR_007891)【63】、tRNAscan-SE(tRNAscan-SE,RRID:SCR_010835)【64】和RNAmmer【65】鉴定了207个microRNAs,34个核糖体RNA,173个转运RNA(tRNA)和1,173个小核RNA。进一步评估了Aquilaria sinensis基因组中非编码RNA的平均长度、总长度及其比例(补充表S13)。此外,预测的基因中有48.61%(14,197个)得到了Iso-seq转录本的支持(补充表S14)。
基因家族鉴定与系统发育树构建
通过保留每个基因的最长转录本,使用Aquilaria sinensis基因组和12种代表植物基因组,包括G. hirsutum(ASM98774v1)、A. thaliana(TAIR10)、T. cacao(Cirollo_cocoa_geneoe_v2)、Cephalotus follicularis(Cfol_1.0)、Citrus clementina(Citrus_clementina_v1.0)、Cucurbita pepo(ASM280686v2)、Eucalyptus grandis(Egrandis1_0)、Glycine max(Glycine_max_v2.1)、Helianthus annuus(HanXRQr 1.0)、Populus euphratica(PopEup_1.0)、Quercus suber(CorkOak 1.0)和Vitis vinifera(assembly 12X),使用全对全BLASTP(1e−5截止值,Blast+ v2.3.056)和OrthoMCL版本2.0.9(Ortholog Groups of Protein Sequences,RRID:SCR_007839)【66】构建了全局基因家族分类。BLASTP和OrthoMCL的默认设置被用于分析。在我们的组装中,共有21,955个基因被聚类为13,713个基因家族。基因家族分析还揭示了789个基因家族和7,248个基因在比较中是Aquilaria sinensis特有的(图3a,补充表S15)。其中,Aquilaria sinensis与4种代表植物(锦葵科的G. hirsutum、椴树科的C. olitorius、梧桐科的T. cacao和作为模式植物的Arabidopsis thaliana)共享了9,615个基因家族,而804个基因家族是Aquilaria sinensis基因组所特有的(图3b)。在APG IV系统中,锦葵科、椴树科和梧桐科均属于锦葵目,而瑞香科也被归为锦葵目【67】。
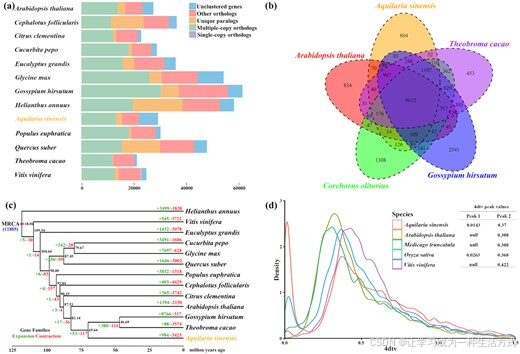
(a) 我们研究的13种植物的基因和基因家族的分布情况。
(b) 展示锦葵目植物中共享基因家族的维恩图,包括沉香树(Aquilaria sinensis)、可可树(Theobroma cacao)、棉花(Gossypium hirsutum)、黄麻(Corchorus olitorius)以及模式植物拟南芥(Arabidopsis thaliana)。
(c) 13种植物间的分化时间估算和基因家族变化。每个节点上的黑色数字表示从现在起估算的分化时间(单位:百万年前)。根节点的蓝色数字(11,885)表示预测的最近共同祖先(MRCA)中的基因家族总数,分支周围的绿色/红色数字表示基因家族的获得/丧失数量。红色节点表示已知的菊类植物和蔷薇类植物的分化时间。
(d) 在选定的基因组组装中,沉香树(A. sinensis)、拟南芥(A. thaliana)、水稻(O. sativa)、紫花苜蓿(M. truncatula)和葡萄(V. vinifera)中4倍简并位点(4dTv)的颠换替代分布。
单拷贝基因
单拷贝基因,或称为孤儿基因,是在物种复制和进化过程中基因组中仅保留一份的基因,这些基因高度保守,通常用于确定物种间的遗传关系和起源。通过Mafft(Mafft, RRID:SCR_011811)【68】对蛋白质序列进行单拷贝基因的比对,然后使用Gblocks(Gblocks, RRID:SCR_015945)【69】过滤比对不佳且高度发散的位点,最终的编码序列(CDS)通过RAxML使用GTRGAMMA模型进行进化分析(RAxML, RRID:SCR_006086)【70】。自展次数为100,H. annuus(向日葵)作为菊科外类群【71】。我们利用PAML的MCMCTREE构建了系统发育树,并通过89个单拷贝基因家族估算了13种植物的分化时间(补充图S5)(参数:clock = 2, RootAge = < 100.6, model = 7, BDparas = 1 1 0, kappa gamma = 6 2, alpha gamma = 1 1, rgene gamma = 2 3.18, sigma2 gamma = 1 1.3;使用菊类和蔷薇类的分化时间[约1.18亿年前]进行校准【71】)。估算Aquilaria sinensis和Arabidopsis thaliana的分化时间为82.14百万年前(95%置信区间,67.63–93.99),A. sinensis与锦葵目物种棉花(G. hirsutum)和可可(T. cacao)的共同祖先的分化时间约为69.64百万年前(95%置信区间,53.18–84.37)(图3c,补充图S6),而我们分析得出的G. hirsutum与T. cacao的分化时间为31.33–69.23百万年前,这与之前的研究结果一致【44, 45】。
基因家族扩展与收缩
特定基因家族的扩展与收缩是植物进化过程中代谢物多样性和物种适应性的重要推动因素【73】。我们使用CAFÉ 2.2(CAFÉ, RRID:SCR_005983)【74】对Aquilaria sinensis基因组中的同源基因家族的扩展与收缩进行分析,使用默认参数。我们比较了13个物种中11,855个基因家族,推测出Aquilaria sinensis基因组中53个基因家族扩展,117个基因家族收缩(图3c,补充表S16)。我们使用Blast2GO(B2G,RRID:SCR_005828)对本体分类(GO和KEGG术语)进行富集分析。扩展的基因家族参与了植物昼夜节律、三羧酸循环、丙酸代谢、核糖体生物合成以及氨酰-tRNA生物合成等途径(补充表S17和图S7),而收缩的基因家族则与淀粉/蔗糖代谢、倍半萜类和三萜类生物合成及亚油酸代谢途径相关(补充表S18和图S8)。
4DTv分布
我们使用MCScanX【75】鉴定了共线区域,选择了每个基因的最长转录本用于分析。在基因家族分析中,BLASTP结果中的前5个互作匹配用作输入。仅包含超过5对基因对的共线性片段被用于计算4倍简并同义位点(4DTv)。通过MUSCLE【76】进行成对序列比对,并对4DTv值进行了修正,以纠正同一位点发生的多次颠换。基于4DTv分布,在Aquilaria sinensis基因组中显著观察到了大量的基因重复事件,这与Arabidopsis thaliana、Medicago truncatula和Vitis vinifera的情形不同(图3d)。


















 357
357

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









