










Gene function exploration and engineering strain library construction from a synthetic biology perspective
合成生物学视角下的基因功能探索与酵母工程菌株文库构建

摘要
合成生物学作为一门通过设计、构建和改造生物系统来实现其特定功能的学科,被广泛应用于生物制造、环境保护和药物合成等领域。基因功能的系统性探索和工程菌株文库的构建是推动合成生物学发展的重要手段。本研究重点介绍了不同酵母文库在合成生物学中的构建方法及其应用前景。随着基因组测序和高通量技术的快速进展,酿酒酵母和裂殖酵母等微生物文库在系统性研究中发挥了关键作用。基因缺失文库、过表达文库、转座子插入文库等多种类型的酵母文库为基因组合优化和代谢路径设计提供了重要工具,促进了代谢工程和合成生物学的创新应用。这些文库在工业生产中支持高产菌株的构建,如用于生物燃料和化学品的高效生产;在环境领域,通过基因改造筛选,生成具备污染物降解能力的菌株,为生态修复提供解决方案;在药物合成方面,文库帮助构建高效合成药用化合物的菌株,推动生物制药的发展。然而,当前文库构建和应用仍面临诸如构建成本、基因组编辑的精确度及筛选效率等技术瓶颈。未来,自动化、数字化和新型筛选技术的进步有望突破这些瓶颈,推动酵母文库的快速构建和高效筛选,从而加速合成生物学在可持续发展和生态工程中的应用。
1 背景介绍
1.1 合成生物学与文库
合成生物学是一门跨学科的研究领域,旨在通过设计、构建和优化生物系统来实现预期功能。其核心理念是将工程学的原则应用于生物学,通过设计新的生物元件、组装新的基因线路以及改造现有生物系统来调控细胞行为,进而推动生物制造、环境保护、能源生产等领域的技术创新。合成生物学不仅涉及基础的基因功能解析,还致力于通过基因的工程化改造实现其在代谢工程、药物开发、绿色能源及生物降解等领域的广泛应用潜力。通过工程化微生物系统,合成生物学能够快速、系统地探索和改造微生物的代谢途径,解析基因的功能与相互作用,从而推动新的生物技术解决方案的产生。
在合成生物学的研究中,工程菌株文库作为一个多功能的创新工具,通过对微生物基因组的系统性修饰,构建一系列包含各种基因变体的菌株。随着20世纪90年代以来模式生物基因组测序的完成及高通量技术的飞速发展,功能基因组学与系统生物学进入了全新的时代[1]。海量的基因组数据不断积累,解析这些基因功能的系统化工具应运而生,文库成为重要的解决方案。这些文库在基因功能的研究中发挥着重要作用,特别是在全基因组范围内系统性地探索基因的功能、相互作用及其在生物网络中的作用。同时,文库也为合成生物学提供了丰富的构建模块和设计工具。通过对基因和调控元件的工程化改造,合成生物学能够实现生物系统的“自下而上”设计,推动在代谢路径优化、药物开发、生物燃料及高附加值化合物生产等领域的广泛应用[2-6]。然而,基因功能探索仍面临许多挑战,包括基因编辑效率、文库构建成本、表型筛选的高效性和精确度等问题。随着技术的不断进步,如何克服这些瓶颈,提高文库构建的速度和精准度,已成为当前研究中的关键需求。
1.2 酵母作为模式生物的重要性
酵母,尤其是酿酒酵母(Saccharomyces cerevisiae)和裂殖酵母(Schizosaccharomyces pombe),作为经典的模式生物,在合成生物学中扮演着重要的角色。酿酒酵母是第一个完成全基因组测序的真核生物[7],其基因组结构相对简单,却保留了大多数真核生物的基本细胞功能。酿酒酵母基因组中约30%的基因与人类疾病相关基因具有同源性[8],因此,酵母成为研究这些同源基因功能的重要工具。此外,酿酒酵母在代谢工程领域的广泛应用,使其成为合成生物学中不可或缺的平台。裂殖酵母则在细胞周期控制、异染色质组织、RNA干扰等方面与酿酒酵母存在显著差异,且在多个方面与多细胞动物的细胞过程更加相似[9-11]。因此,裂殖酵母也成为研究真核生物细胞机制的重要工具。酵母被广泛应用于生物发酵、燃料生产、蛋白合成等工业领域。在传统发酵工业中用于生产酒精、面包、酱油等食品和饮料。已经筛选出了可以利用木糖、葡萄糖等多种廉价碳源,生产乙醇等生物燃料[12]。此外,基因工程技术被用于开发新的代谢途径,生产多种有价值的化学品,如维生素、香料和疫苗、胰岛素等药用分子。
1.3 酵母文库构建的意义
酵母文库的构建为基因功能的解析和合成生物学的应用提供了强大的支持。不同类型的酵母文库,如基因缺失文库、过表达文库、荧光蛋白标记的开放阅读框(open reading frame, ORF)文库、转座子插入文库、RNA干扰文库和表达谱文库等,能够通过高效的筛选系统对基因进行全面功能探索。在文库包含的菌株集合中,每个菌株中不同的基因以相似的方式被修饰,从而实现平行的、无偏倚的全基因组水平的基因功能探究[13]。这些文库不仅帮助揭示基因在细胞中的功能,还能在代谢工程中用于优化代谢路径,提升酵母对不同底物的转化能力。通过基因过表达或基因敲除文库的构建,研究人员能够有效改造酵母,用于合成高附加值化合物,如药用分子和天然产物。例如,通过全基因组文库筛选,研究人员能够发现对特定底物(如木质纤维素、葡萄糖等)的发酵能力强的酵母菌株,从而提高生物燃料的生产效率[12]。一些酵母株被改造用于合成药物成分,从葡萄糖直接合成具有抗氧化、抗炎和抗微生物的特性黄酮类化合物[14],通过全基因组分析可以找到提高合成效率的关键基因。再如,有研究发现酵母通过表达特定的酶,能够高效地水解聚乳酸(Polylactide,PLA),从而促进其回收与绿色化处理,有助于推动可持续发展,同时展现了文库在新型生物降解材料开发领域的潜力[15]。
2 酵母文库的构建方法
2.1 经典方法
2.1.1 传统的基因敲除和过表达策略
酵母单基因缺失文库[16-18]中,每个菌株中被删除的目的基因ORF都通过同源重组法被替换成了一个卡那霉素抗性筛选标签(Kanamycin Modular eXpression,KanMX),并且两侧有一对长度为20bp的序列各不相同的标签,被称为“分子条形码”(barcode)(图1a)。这些条形码能够准确唯一地识别每个缺失突变体,这使高通量的定量平行分析成为可能,为全面、快速、经济、便捷地筛查各个生物学方面的基因功能奠定了基础[19-20]。每个基因缺失菌株携带的独特的分子条形码大大简化了在同一实验条件下对多个突变体进行平行分析的过程。例如为了筛选对生长重要的基因,通过一次实验,将所有单基因缺失菌株混合培养,并在生长过程中定期采集样本。从这些样本中提取基因组DNA,通过扩增与每个单基因缺失菌株相关联的分子条形码。即可根据该基因的丰度判断此基因对生长的重要性。对生长越重要,相应缺失菌株的分子条形码在培养过程中就减少得越快。因此,所有目的基因都可以在一次实验中被识别出来,并按照它们对适应度的相对贡献进行排序(图1b)。
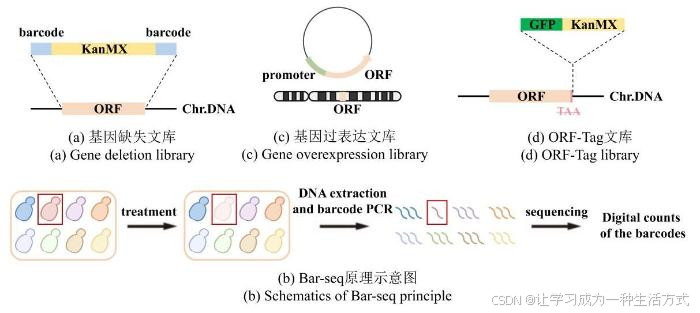
图1 经典文库构建方法示意图
(本图由biorender绘制)
(a)基因缺失文库中,目的基因ORF(橘色片段)被替换成卡那霉素抗性筛选标签KanMX(黄色片段),两侧伴有“分子条形码”barcode(蓝色片段)。Chr.DNA:染色体DNA。(b)Bar-seq原理示意图。在特定条件处理下,目标菌株(红色)的生长量相对低(淡粉色),表现为测序时相应barcode的读数量相对低(红色线条比其他颜色少)。(c)基因过表达文库中,目的基因ORF(橘色片段)除了在基因组上正常表达之外,还额外在质粒上由通用型启动子(淡绿色片段)驱动表达。(d)ORF-Tag文库中,在删除终止密码子的目的基因ORF(橘色片段)C端插入荧光蛋白GFP(深绿色片段)和筛选标签KanMX(黄色片段)。Chr.DNA:染色体DNA。
全基因组过表达文库通过将一系列携带额外目的基因拷贝的质粒载体与强启动子相结合(图1c),强制性提高每个基因的表达水平,来研究基因在细胞生长、代谢通路及应激反应中的作用。质粒上的启动子通常是诱导性的,如gal1启动子[21]或nmt1启动子[22],他们在半乳糖碳源或去除硫胺素的条件下可以诱导有该启动子的基因强表达,这使得研究人员能够在特定条件下调控基因表达从而观察表型变化。
ORF-Tag文库通过在基因的C端或N端插入荧光标签来对目标蛋白进行标记。它有两种模式,一种是在各个基因的原生位点进行修饰(图1d),另一种则是在一个异位位点上由额外表达融合蛋白。前者保证了目标蛋白的表达水平和模式受到的干扰最小;后者往往涉及基因过表达,一些在正常状态下不表达的基因产物也能够通过诱导表达在细胞内进行定位。但与此同时,融合蛋白的非生理表达可能导致其定位错误,对于一些剂量敏感型蛋白还可能造成细胞内分子成分之间平衡的扰动或紊乱,影响细胞正常生理活动。
2.1.2 交配和选择
2001年,Tong等人开发了SGA(synthetic genetic analysis)方法用于系统构建双基因修饰(敲除或过表达)文库[23],该方法利用自发的交配途径代替了冗长的转化过程。以构建酿酒酵母单倍双突变体为例(图2),在缺氮条件下诱导两种相反交配类型(MATa和MATα)的细胞交配,这些母本菌株包括以下三个特征:①它们分别在各自的突变位点携带不同抗性标签(KanMX和NatMX)用于筛选双突变体子代(图2a)。②在任一母本菌株中携带负筛选型突变(如can1Δ、lyp1Δ等),用于在选择性培养基上反筛选二倍体(图2b)。含有野生型can1基因的单倍体细胞或二倍体细胞会摄入有毒的刀豆氨酸(canavanine),从而被杀死,而can1Δ突变体无法将刀豆氨酸运转入体内,因此能够存活。③在某一细胞型的母本菌株中,构建另一细胞型特异性启动子与营养缺陷筛选标签的表达盒,用于选择任一性别的单倍体子代细胞(图2c)。如在MATa细胞中构建MATα特异型启动子STE2pr驱动his5基因的表达盒,只有当细胞发生交配产孢后,STE2pr-his5才能在MATα子代细胞中表达从而允许其在组氨酸缺陷型培养基上生长;同理,在MATα细胞中预先组装STE3pr-lue2表达盒,由于STE3pr只能在MATa细胞中表达,因此可以通过亮氨酸缺陷型培养基筛选MATa子代细胞。除了使用细胞型特异性启动子以外,Krogan课题组还在裂殖酵母中开发了一种特定单倍体的偶联筛选策略PEM(pombe epistasis mapper),他们通过将一个显性致死的抗性基因(cyhS)“镶嵌”在交配位点mat1附近,使得某一单倍体表型与抗性基因的表达偶联,在添加环己胺(cycloheximide)的培养基上实现对这一单倍体的反筛[24](图2d)。根据双基因缺失菌株相对于任一单基因缺失菌株的生长情况,可以判断两个基因之间发生的负向或正向相互作用,揭示基因间的冗余或旁路抑制。
图2
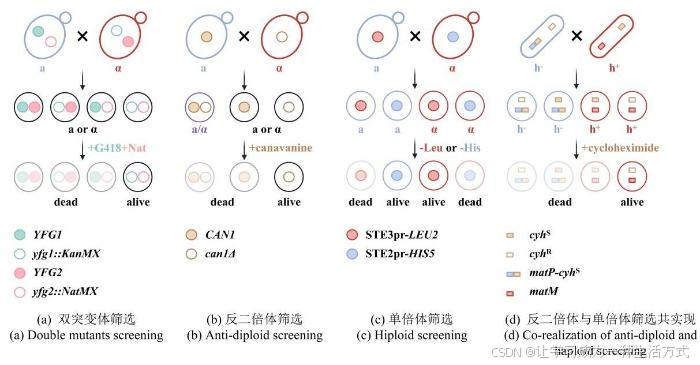
图2 SGA与PEM方法原理图
(本图由biorender绘制)
(a)双突变体筛选策略。两种交配型的细胞分别在各自的突变位点携带不同抗性标签KanMX(绿色空心圆点)和NatMX(粉色空心圆点)用于筛选双突变体子代(黑线框细胞)。YFG (your favourite gene) :目的基因。(b)反二倍体筛选策略。含有野生型CAN1基因的单倍体细胞(蓝色细胞,a型)或二倍体细胞(紫色细胞)会摄入有毒的刀豆氨酸(canavanine),从而被杀死,而can1Δ突变体无法将刀豆氨酸运转入体内,因此能够存活(黑线框细胞)。(c)单倍体筛选策略。在某一细胞型的母本菌株中,构建另一细胞型特异性启动子与营养缺陷筛选标签的表达盒,用于选择任一性别的单倍体子代细胞。例如在a型细胞(蓝色)中,携带只能在α型细胞中表达的STE3pr-LEU2基因线路(红色实心圆点),只有细胞交配使STE3pr-LEU2基因线路存在于α型细胞中时,该细胞存活(含有红色实心圆点的红线框细胞)。(d)反二倍体与单倍体筛选共实现。通过将一个显性致死的抗性基因cyhS(棕色实心方块)“镶嵌”在裂殖酵母交配位点mat1(在蓝色的h-细胞中为蓝色方块表示的matP)附近,使得某一单倍体表型与抗性基因的表达偶联(matP-cyhS )。
基因缺失文库是研究非必需基因全基因组功能分析的宝贵资源,而显然,必需基因敲除的单倍体菌株是不可存活的,这阻碍了其在后续多种不同条件下的表型分析。但必需基因往往对细胞更加重要,与非必需基因相比,酵母必需基因在其他生物体中表现出更多的同源性[25],并参与了更多的蛋白质-蛋白质相互作用[26-27]。为了研究必需基因,研究者们已经尝试构建了许多特定的突变体,也称为条件等位基因,来对必需基因进行遗传分析。这些突变体能够以类似变阻器开关的形式在某一条件下保留基因功能,而另一条件下缺乏基因功能,甚至在某种半许可的条件下表现出部分的不完整的功能以供进行多角度分析。此外,兼容了SGA筛选标记和独特条形码的必需基因条件等位基因文库能够轻松地与基因缺失文库进行交叉互作并实现高通量。
条件等位基因主要有三种模式。①温度敏感型(Temperature Sensitive,TS)突变体[28](图3a)。该等位基因表达的蛋白在正常或略低的培养温度下正常发挥功能,而在较高温度下因为蛋白折叠问题而丧失功能。温度敏感突变等位基因的生成方式包括随机诱变[29-30]、基于蛋白质结构预测技术的理性设计与定向诱变[31-32]、在宿主蛋白内部插入温敏型内含肽[33]等。温度敏感突变体亦可携带分子条形码,从而便于高通量平行分析,通过“plasmid shuffle”[34]或“diploid shuffle”[30,35]将其整合到宿主中,广泛用于研究必需基因主导的生物过程,如细胞分裂和染色体稳定性[28,30]等。然而,这种方法的缺陷是温度的变化对细胞生理活动的普遍扰动,且已有的必需基因蛋白在温敏条件下仍然可能发挥作用,因此必需基因的功能研究仍不完全。②诱导型启动子替换原生启动子(图3b)。例如,使用四环素诱导型启动子TetO7替换原启动子,实验表明,添加足以关闭该基因表达的强力霉素对整体基因表达几乎没有影响[36]。此外,Charles课题组开发并鉴定了β-雌二醇诱导的YETI文库,每个基因的原生启动子被替换成一个工程化后的雌二醇诱导型启动子,每个基因在其原生位点上能够根据不同雌二醇浓度具有不同的表达水平[8]。由于β-雌二醇不是酵母代谢物或信号分子,因此能够极大程度地避免细胞代谢受到干扰。不同诱导物浓度与相应启动子的结合能够更加精准地调控基因表达水平,实现类似“变阻器”的效果,更好地研究基因剂量效应。③通过mRNA扰动降低蛋白丰度[37-38](图3c)(decreased abundance by mRNA perturbation,DAmP)。此策略通过在在必需基因的3′UTR区插入一个抗生素耐药性标记物,降低目标mRNA的稳定性和丰度,使蛋白质水平在天然转录调控下降低,却足以支持菌株生存能力。对于必需基因来说,即便是基因的低水平表达也可以维持细胞的生存,但仍然能表现出一定的生长缺陷或特定的表型,从而揭示该基因的功能。
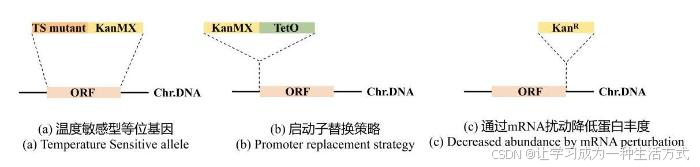
图3 条件等位基因文库示意图
(本图由biorender绘制)
(a)温度敏感型(Temperature Sensitive,TS)等位基因中,目的基因ORF(橘色片段)被替换成相应的温敏突变基因TS mutant(深橘色片段)并携带一个抗性筛选标签KanMX(黄色片段)。Chr.DNA:染色体DNA。(b)启动子替换策略中,携带抗性筛选标签KanMX(黄色片段)的诱导性启动子TetO(绿色片段)被插入目的基因ORF(橘色片段)的起始密码子上游。Chr.DNA:染色体DNA。(c)通过mRNA扰动降低蛋白丰度的DAmP策略中,目的基因ORF(橘色片段)的3’UTR区被插入一个抗性标签KanR(黄色片段)。Chr.DNA:染色体DNA。
实际上,有科学家提出,基因的必需性并不是一个固有的内在属性,而是能够受遗传和环境因素影响而可变的。通过酿酒酵母杂合二倍体基因缺失文库减数分裂产生一个必需基因缺失文库,使其在适宜的条件下发生适应性进化,而后评估该文库中约1,000个必需基因缺失菌株的存活程度。结果表明,文库中约9%的致死性突变可以被适应性进化所克服。基因必需性的重新审视和定义与突变细胞的存活性和进化能力将有助于选择与耐药性风险较低的相关治疗靶点[39]。
2.2 现代技术
2.2.1 转座子介导的文库构建
相较于缺失突变文库中基因功能的完全丧失,随机插入突变可能会以不同程度消除、减少或改变基因的功能。在编码序列中的插入可以产生没有功能或功能改变的截断蛋白。在基因的5 '和3 '区域插入可以通过损害启动子功能和mRNA稳定性来改变蛋白质表达水平。例如,Hermes转座子已经被发现可以有效地插入S. pombe和S. cerevisiae基因组[40-43]。Hermes转座子的两端各有一段末端倒置重复序列(terminal inverted repeats, TIRs),转座酶通过识别TIRs来剪切转座子并将转座子DNA整合到新的位置。
基于转座子插入文库,衍生了一种高通量的全基因组研究方法,称为转座子插入测序TN-seq[44-45]。它的工作原理如下:当一个足够大、包括足够多的单个转座子插入菌株文库被构建后,我们就可以通过其在基因组上插入位点的偏好性来识别出必要的基因或区域。因为能够被测序的细胞必然是能够存活的,因此其被插入位点都是非必需的。接下来,若将转座子插入文库置于选择压力下,并重新测序得到插入频率从初始生长条件到生长条件改变后的变化,即可得到与选择压力相关的基因区域。TN -seq文库中每个基因突变密度的大幅增加为每个基因对特定表型的贡献提供了更多信息。同时,它将减少由于只分析一个基因突变而导致假阳性或假阴性的可能性。例如,TN-seq可用于筛选裂殖酵母中维持孢子活力和健康所需的基因[46]。Sarah课题组首先在一个h90菌株中生成了一个转座子插入文库,待细胞交配产孢后,分离出孢子并通过PCR扩增转座子相关DNA后测序。通过比较在诱导有性生殖之前和之后转座子插入某一相同基因的频率,来获取与有性生殖密切相关的基因集。他们发现许多已知的减数分裂相关基因中的插入表现出明显的频率减少,因为在减数分裂相关基因位点有插入就无法进行减数分裂,这个结果也同时验证了TN-seq的有效性。作为对照组,他们分析了基因组上基因间区域的插入频率,它们经对数调整后的平均比率略低于零。相反,那些与基因间区具有显著不同的倍数变化分布的基因则被定义为有性生殖基因,为后续的验证研究提供方向。
2.2.2 CRISPR/Cas9技术
毋庸置疑,CRISPR/Cas系统已经成为一种成熟高效的基因组编辑工具,其中应用最广泛的系统之一是CRISPR/Cas9系统[47-48]。CRISPR/Cas9系统通过工程化的单链向导RNA (Single guide RNA,sgRNA) 引导Cas9核酸酶切割基因组,造成DNA双链断裂[49]。当供体DNA存在与基因组DNA同源序列时,同源重组修复机制将会促使供体DNA与断裂部分DNA发生交换,从而完成基因组修复并实现供体DNA的插入。将Cas9蛋白点突变使其失去核酸内切酶活性,失去活性的Cas9称之为dead Cas9(dCas9)[50]。dCas9虽然失去了切割DNA的能力,但仍然可以在sgRNA的引导下与特定的DNA序列结合。通过将dCas9与转录效应因子(如转录激活因子或抑制因子)融合,可以实现对目标基因的转录调控。目前,CRISPR系统已被应用于实现基因敲除、整合、转录干扰和转录激活[51],几乎所有代谢工程中的基因操作都可以通过CRISPR/Cas9系统完成[52]。
为了更合理地构建菌株文库,目前已经建立了基于高通量基因编辑技术的随机组合自动化多重基因工程(Multiplex automated genome engineering,MAGE)[53]。该方法利用在基因组中存在多个位点(100个以上)的逆转录转座子δ[54],将其末端的重复序列设计为供体DNA的同源臂,结合CRISPR/Cas9系统在δ序列中引入双链断裂,大大提高整合效率。此外,寡聚核苷酸介导基因组工程(Yeast Oligo-Mediated Genome Engineering,YOGE)[55]和真核生物多重基因组工程(Eukaryotic multiplex genome engineering,eMAGE)[56]也被应用于酿酒酵母的多位点靶向工程化改造中。Keasling课题组提出了一种名为CasEMBLR的方法,通过结合体内DNA组装和Cas9介导的多位置整合,实现酿酒酵母基因组的无缝、无标记和稳定基因组工程[57]。Liu等基于CRISPR系统设计了一种组合重编程基因表达以操纵复杂表型的方法(Multiplex navigation of global regulatory networks,MINR),并构建了MINR文库[58]。MINR 文库是针对 25 个调节基因(例如,转录因子、抑制因子和共激活因子)设计的 ,包含总共 43,020 个与不同应激反应相关的特定突变。为了证明 MINR 文库在工程复杂表型中的效用,他们通过乙醇和/或葡萄糖耐受性实验,成功筛选到了乙醇和/或葡萄糖耐量提高,且乙醇的生产浓度也比野生型菌株提高了2倍的特异性突变体,展示了其在工业应用中的巨大潜力。
2.3 文库构建的挑战与限制
当然,这些文库在发挥重要作用的同时也存在一些由于基因组的复杂系统性带来的局限性。例如基因组上存在基因冗余:许多基因在功能上相似,导致文库中存在冗余克隆,这使得在筛选和分析过程中难以确定特定基因的独特功能[59-60]。在克隆和测序过程中,基因组的复杂性可能导致重叠区域难以正确拼接,影响基因组序列的准确性。由于基因组的可塑性,不同个体或株系的基因组存在结构变异,可能导致文库中的某些基因缺失或改变,从而影响研究结果的可重复性[61]。虽然酵母基因组已被研究者们深入的研究,但是仍有很多不确定功能的基因,复杂的基因组结构和未注释基因增加了功能注释的难度,使得研究人员需要投入更多时间和资源进行基因功能分析[62]。
选择合适的载体对成功构建文库至关重要[63]。如果载体不适合目标基因的表达或功能,后续的筛选和分析效果将受到影响。载体的选择不仅要考虑其是否能高效地携带目标基因,还要确保其在宿主细胞中的稳定性和复制能力。载体的质量和特性直接影响到文库的可靠性和功能性。另一方面,不同的表达系统对特定基因的表达效果各异[64]。因此,在选择表达系统时,需要综合考虑目标基因的来源、功能和后续应用。选择不当可能导致基因无法有效表达或失去功能,从而影响后续的实验和应用。设计有效的筛选策略也是成功构建文库的关键[23]。筛选策略需要针对文库的具体目的,选择合适的筛选标志物或表型指示器。通常,这需要进行多次实验和优化,以确保筛选过程的高效性和准确性。如果筛选条件不够严格或不够灵敏,可能会导致有效候选株的遗漏。
最后,构建文库通常需要大量的时间和资源投入,包括试剂、设备和人力成本,这对于小型实验室或初创公司来说可能是一个重大限制。如何平衡文库构建的复杂性和资源的可用性,以确保实验的可行性和有效性,仍是一个挑战。克服这些挑战需要通过优化实验设计、改进技术和方法,以及合理选择材料,从而提高基因组文库的构建效率和质量。
3 酵母文库在研究中的应用
3.1 基因功能研究
3.1.1 基因相互作用的解析
将基因型与表型联系起来,并从这些关联中推断出基因-性状的因果关系,对于理解复杂性状的分子机制至关重要[17,25]。在系统性文库被构建之前,经典的遗传学方法主要包括基因足迹分析[65-66](genetic footprinting)和随机突变(random mutagenesis)。遗传足迹分析是通过转座子随机插入基因组获得突变体库;随机突变则是通过易错PCR(error-prone PCR)或物理和化学诱变等方法来造成基因序列改变。这些传统的遗传筛选方法存在局限性,通常会遗漏某些突变的表型。相比之下,定向的基因缺失文库表现出了它的显著优势:首先,作为一种反向遗传方法,突变与表型是一一对应的,每个基因的敲除都可以清晰地关联到特定的表型。研究人员无需鉴定负责某一突变表型的基因,而是能够直接通过观察表型来推断该基因的功能。其次,突变表型反映了该基因功能的完全丧失,这很好地避免了基因功能丧失不完全而导致其重要性被掩盖或产生模糊的表型。此外,与随机突变相比,文库中的基因组突变是“饱和”的,这意味着每个基因的功能都可以在文库中被系统性地研究,而不会因随机突变的覆盖不全而遗漏。
除了富营养生长相关功能基因筛选,单基因缺失文库的条形码测序技术(Bar-seq)被广泛应用于分析细胞在不同组分的培养基[25,67-68]、不同环境压力[67,69](高渗透压、高盐碱、高温)、不同化学物质[70-73](抗生素、毒素、生物活性抑制剂)等一系列条件下的生长关键基因,或在特定生理活动[70,74-76](减数分裂、产孢)中的关键基因。与最小基因组的鉴定和合成真核基因组密切相关的必需基因集也是通过此文库筛选出来的。研究者们发现18.7%的基因对酿酒酵母在葡萄糖富培养基中的生长是必需的,而在其余存活的单倍体单基因缺失菌株中,另有15%表现出生长缓慢,主要包括核糖体蛋白和线粒体功能的编码基因[25]。在裂殖酵母中,在YES富培养基上生长所需的必需基因占比为26.1%(1260/4836)[18],杜立林课题组通过比较分析裂殖酵母的单基因缺失文库在YES、EMM和添加赖氨酸的EMM培养基中的生长情况,发现了多种在最小培养基中表现出特定生长缺陷的营养缺陷突变体[67]。再例如,多伦多大学的Charles Boone课题组使用82种不同的化合物对酿酒酵母单基因缺失文库进行处理和筛选,获得了一张庞大的化合物-基因互作图谱,并通过二维层次聚类进行可视化[73]。该图谱有助于揭示对其他化合物的未知作用模式,或通过基因在不同处理条件下的聚类来预测其未知功能,对于合成生物学中合成各种化合物的通路分析,建立有用的合成细胞工厂和底盘都有着重要的作用。
Bar-seq中所有突变体被混合在同一培养环境中进行生长,这可能导致不同菌株之间的竞争效应被放大,从而影响对某些基因功能的评估。此外,Bar-seq方法噪声较高,尤其是在检测低丰度突变体时,其灵敏度有限,容易导致这些突变体的信号难以被准确捕捉[67,77]。相比之下,单独培养并观察每个缺失突变体在特定条件下的表现虽然工作量较大,但这样严谨细致的筛查可以避免大多数遗漏,因此许多研究者仍然选择单个菌体筛查[12,78-80]。另外,一些难以通过群体生长状态来量化的表型(细胞形态[80-83]、交配表型[84]等)也需要对每个菌株进行单独检查。举例来说,在筛查与DNA损伤反应(DNA Damage Response,DDR)相关的基因时,Hartsuiker课题组使用不同浓度的DNA损伤剂对裂殖酵母单基因缺失文库中的2662个单倍体菌株进行逐一处理和观察,共发现229个对这些试剂敏感的突变体,它们与DNA复制与修复、DNA损伤检查点、染色质结构密切相关[85]。同时,杜立林课题组[67]和吕红课题组[86]分别对一个更加完整的单基因缺失文库进行bar-seq和平板筛查,对这个基因集进行了多重验证和补充,为构建一个更全面的DDR网络做出了贡献。
通过SGA方法,Charles课题组已经绘制了一张酿酒酵母全局基因互作网络图谱,可视化了全局基因互作图谱的相似性网络,并利用功能富集空间分析(SAFE,spatial analysis of functional enrichment)识别与特定功能属性相关的密集网络区域[87]。全局基因互作相似性网络展示了一个有组织和功能全面的细胞功能视图,GO富集条目被映射到17个独特的子网络区域,覆盖了全网络上的1300多个基因。属于相似生物过程的基因倾向于共享相同的基因相互作用,编码在同一途径或复合物中共同发挥作用的蛋白质的基因会显示出相似的基因相互作用谱。通过未知基因在相似性网络图谱中功能富集区域的定位,研究者能够预测基因功能。
3.1.2 蛋白质定位与功能的探索
全面了解蛋白质在细胞微环境中的定位对于理解它们的功能和相互作用至关重要,这需要分析细胞内的全部蛋白质。确定模式生物的基因产物的功能是了解它们在后生动物中的功能的重要一步,并为更完整地理解细胞过程和途径奠定基础,从而成功开发对抗疾病和合成生物学发展提供方案。早期研究人员对酿酒酵母蛋白定位的大规模分析依赖于转座子介导的随机表位标记和基于质粒的表位标记蛋白的过表达[88-89]。然而,部分开放阅读框(orf)的表位标记可能中断重要的定位信号,而蛋白的过表达可能使细胞内运输机制饱和,导致亚细胞定位异常。ORF末端标记能够很好地规避这些潜在的问题。在2003年,酿酒酵母C端ORF-GFP融合文库就已经被构建完成,该文库包含4156个高于GFP可检测水平的菌株,覆盖了酿酒酵母蛋白质组的75%。每个菌株中的特定融合蛋白在其原生位点被标记和表达,由此保证蛋白质表达水平和模式受到的干扰最小[90]。他们将这些蛋白质分为22个不同的亚细胞定位类别,该研究首次提供了这一高分辨率、高覆盖率的定位数据集。随着近二十年人工智能的发展,蛋白质定位预测算法的不断开发和优化推动了大规模图像数据集定量分析的进步[91]。计算方法通过以无偏倚的定量形式提取形态学信息,使数百万单细胞图像的高通量识别与分析成为可能。理想情况下,计算工作流可以将单个细胞和蛋白质映射为鲁棒的数值来表示,从而以客观的方式分析细胞的空间组织。例如,Brenda课题组创建了一个自动化算法来量化上述ORF-GFP文库中蛋白的丰度和定位,并进行了可视化[92]。他们生成了一个体现全局蛋白质组功能信息的可视化丰度定位图谱ALM(abundance localization map),具有共同定位特征的蛋白质能够被聚类。此外,他们还生成了另一个网络来可视化不同亚细胞隔间之间共享定位模式的比例,以及共享蛋白的平均丰度。这些定位和丰度数据是定量的,因此为多种类型的比较研究、单细胞分析、建模和预测奠定了基础。
在裂殖酵母中最早的ORF-Tag文库可以追溯至2009年,Hayashi等人构建了一个ORF-YFP文库,其中每个菌株除了表达各自的原生基因之外,额外在一个异位位点(leu1)上由诱导型启动子nmt1驱动表达融合蛋白,这个文库覆盖了近90%的蛋白质组[22]。Matsuyama等人在基因原生位点构建了一个不完整的ORF-GFP文库,文库中涉及的1630个基因主要编码核结构和细胞周期控制相关蛋白,补充提供了关于细胞蛋白定位和丰度的信息[93],但裂殖酵母全基因组蛋白标记文库仍是空白。
3.2 工业菌株的适应性进化
合成生物学中常用的工程菌株的工业化应用往往涉及多基因协同作用的复杂表型,在基因组规模上对代谢途径进行编辑或重排,获得具有优化或额外添加的性状的细胞株,如异源产物生产、底物高耐受性等[94-95]。适应性工程通过在培养基中增加压力并连续转移培养,来分离具有高耐受性的进化菌株。适应性工程虽然有效,但却非常耗时,因为期望突变的出现是不常见的,而且常见的突变大多数都是有害的或中性的。随着自动化设施和基因组编辑技术的发展,多种高通量筛选方法被开发,以高通量形式在基因组水平上修改细胞生理功能来重建细胞工厂,为菌株生长和产物积累提供最佳条件,最终提高细胞工厂的生产性能[96-98]。由于合成产物的复杂性状的工程往往需要许多基因的同时调节,因此在基因组规模上有效地产生多重遗传多样性的新的方法是非常必要的。
Keasling课题组通过共转化携带不同gRNA质粒和相应的供体DNA片段,开发了用于酿酒酵母基因组多基因敲除的CRISPR/Cas9系统,最多可以同时对5个不同的基因组位点进行敲除[99]。通过多重基因组编辑的组合对酿酒酵母中的甲羟戊酸水平进行了探索性分析,确定了一株在没有引入任何外源基因的情况下甲羟戊酸产量比野生型菌株提高了41倍的工程菌株。结合CRISPR/Cas9系统,Zhao课题组设计了一个自动化的系统来整合全基因组筛选和多重优化[53,95]。他们通过将一个全基因组的cDNA文库正向或反向插入一个转录单元中,构建了基因过表达或沉默的供体文库。将该文库转入整合了RNA干扰途径的酵母细胞中,目的片段利用逆转录转座子的长末端直接重复序列δ整合在基因组中。在全长cDNA分子沿正义方向转录时,基因过表达;在全长cDNA分子沿反义方向转录时,全长反义RNA通过RNA干扰导致基因沉默。此外,他们借助CRISPR-Cas9系统提高整合效率。该策略被应用于醋酸高耐受性菌株的筛选,他们成功得到了高于任何先前水平的醋酸抗性,且大大减少了时间和人工工作量。全基因组测序数据表明,该菌株至少整合了26个基因的过表达或沉默,表明该策略在高效创建多重基因多样性方面是有效的。有趣的是,基于RNA干扰途径的基因沉默相比于基因序列删除来说是不完全的,对于不同基因不同区段的沉默可能会获得较弱的效果,甚至可能得到相反的表达模式。而对于同一种特定的表型,某个基因功能的丧失或增强都有可能促进表型[58]。因此菌株的定向进化不仅需要多位点同时产生突变,也需要更加多样化的干扰策略,例如基因不同程度的减弱或过表达[53],CDS区域的饱和突变[52],UTR区的修饰[100]等等。
4 未来展望
4.1 新兴文库构建技术
4.1.1 基于自动化的高通量筛选技术
高通量筛选技术的进步使基因组文库的构建和筛选变得更加高效和精准。传统的筛选方法通常耗时且劳动密集,而高通量技术通过自动化和并行化的方式,大幅度提高了筛选的速度和效率,并增强数据的可重复性和可靠性[63,101-102]。现代高通量筛选平台能够在短时间内处理成千上万的样本,从而显著缩短文库构建的周期。数字化管理系统帮助研究团队有效记录和跟踪实验数据,集中管理样本信息、实验步骤和结果,提升实验的透明度和可追溯性。此外,微流控技术的应用使得在微米尺度上控制液体流动变得更加灵活,有助于快速筛选出目标细胞,节省大量实验时间。结合自动化液体处理系统,研究人员能够实现高通量的实验设计,确保样本处理的一致性和重复性[103]。多重检测技术的引入,如荧光、质谱和基因组测序,能够同时检测多个目标,提高筛选的灵敏度和特异性。随着计算技术的发展,数据分析方法也日益先进,使研究人员能够快速处理和解析大量筛选数据,加速文库构建后的表型分析进程[17,91]。这些技术的融合,正推动基因组文库构建技术的不断发展,使其更加高效、灵活和精准,为基础生物学和应用研究提供了强大的工具和平台。
4.1.2 合成生物学的工程化方法
建立一个文库是劳动密集成本较高的工作,为了实现快速构建文库切换的可能,Maya课题组提出了一种ORF-Tag文库的策略,被称为SWAT(SWAp-Tag)[104]。该方法基于一个特殊的底盘受体文库,其中每个菌株都在基因组上的某一位置含有可被交换的受体模块,通过SGA方法将受体文库与含有新标签的供体菌株杂交并筛选,从而快速有效地获取一个全新的文库。这种方法的一个关键优点是,受体模块的任何部分(启动子、选择标签、甚至整个受体模块)都可以被任何其他部分取代;替换整个受体模块为荧光标签,能够恢复天然的基因调控模式,达成无缝标记。作为方法验证,他们在酿酒酵母中构建了一个与内膜系统蛋白相关的N端ORF-GFP文库,首次可视化了600个内膜系统蛋白,并对C端文库进行了补充和修正,同时还发现了n端和c端标记对蛋白定位的影响情况。由此可见,构建各种互补的文库来相互映证和修订是很有必要的。
4.2 新型文库的构建
除了前面提到的双基因缺失文库,更加复杂的多维因素(基因或环境)互作文库也已被报导。三基因缺失互作筛选或双基因缺失突变体在不同环境条件下的筛选能够进一步补充揭示基因的隐藏功能[105-106]。作为一个应用实例,Charles课题组利用三基因相互作用分析对来自于全基因组复制的同源基因(paralog gene)的功能冗余程度和进化轨迹进行了探究。通过赋予一个三基因相互作用分数来反映同源基因对的功能重叠程度。理论上,高度分化的基因对应该表现出相对较多的特异性负基因相互作用。相反,功能重叠的基因对应该偏向于更多的三基因相互作用,展现出较少的特异性相互作用。此外,将同源三基因相互作用映射到全局双相互作用相似度网络上,可以对以前未确定特征的同源基因进行功能注释。这项研究填补了功能冗余和进化轨迹研究中的空白,使得我们对同源基因的功能重叠和相互作用有了更深入的理解,进而为后续的基因功能研究和生物工程应用提供了新的视角和思路。
4.3 文库构建在合成生物学中的应用前景
4.3.1 人工生命的构建
酵母基因组文库在人工生命构建中发挥着重要作用,主要体现在基因资源的丰富性和实验效率的提升。通过这些文库,研究人员能够筛选出与代谢、信号传导和基因调控相关的关键基因,深入了解生命的基本机制。“人工合成酿酒酵母基因组”计划旨在设计和构建一个完全人工合成的酵母基因组[107-111],此目标的实现将意味着人类可以从修补少数基因到从头设计和构建整个基因组,必定是一个激动人心的里程碑。单条染色体酿酒酵母的人工构建不仅为为研究真核生物的染色体结构和功能进化提供了一种替代方法,而且对于端粒生物学、着丝粒生物学、减数分裂重组以及核组织和功能之间关系的未来研究具有相当大的价值[112]。酵母基因组文库为基因工程和合成生物学提供了重要的支持,使研究人员能够进行基因组编辑和合成新的基因线路,为人工生命的构建提供了坚实的基础。这些进展不仅加深了对生物功能复杂性的理解,也为合成生物学的应用提供了新的可能性。
4.3.2 对生态与环境的影响
酵母基因组文库在生态与环境研究中的潜力也日益受到关注,其作用主要体现在以下几个方面。首先,这些文库为研究微生物在生态系统中的功能提供了丰富的基因资源,使科学家能够深入分析与环境适应性、营养循环和生物相互作用相关的关键基因,这有助于揭示微生物在维持生态平衡中的重要角色。其次,酵母基因组文库在环境生物修复中具有重要应用价值。例如,筛选对廉价清洁能源利用率高的菌株以用于目标产物合成[113]。未来,通过筛选具备特定降解能力的菌株,研究人员可以开发新型的生物修复策略,以有效去除土壤和水体中的污染物。此外,酵母基因组文库的应用还促进了合成生物学在生态研究中的发展。利用这些工程化酵母设计出具有特定生态功能的微生物,如碳捕集、氮固定和有害物质降解,将为应对气候变化和生态退化等全球性问题提供了新的解决方案。最后,酵母基因组文库的研究能够帮助科学家更好地理解生物多样性和生态系统功能。通过比较不同酵母株的基因组和表型,研究人员可以揭示微生物多样性的形成机制及其对生态系统的影响,从而为保护生态环境和维持生物多样性提供科学依据。


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









