









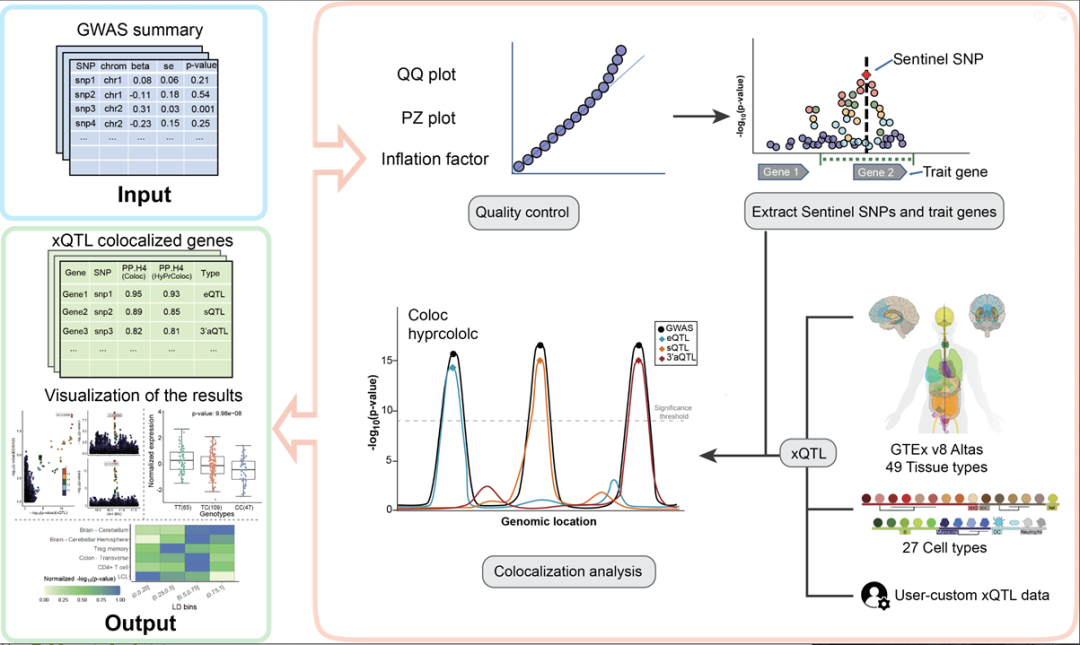
XQTL 共定位分析
XQTLbiolinks 是一个端到端的生物信息学工具,由深圳湾实验室李磊研究团队开发,用于高效地分析公共或用户定制的个xQTLs数据。该软件提供了一个通过与 xQTLs 共定位分析进行疾病靶基因发现的流程,以检测易感基因和致病变异。
github地址:
https://github.com/dingruofan/xQTLbiolinks
0. R包下载与引用
用户可以通过以下代码安装或者引用:
#if (!require("BiocManager", quietly = TRUE)){install.packages("BiocManager")}
#BiocManager::install("SummarizedExperiment") # For windows or linux
#if(!require("devtools")){install.packages("devtools")}
#devtools::install_github("dingruofan/xQTLbiolinks")
library(data.table)
library(xQTLbiolinks)
library(stringr)
library(coloc)1.数据预处理
使用示例文件(GRCh38 版本基因组)的 GWAS 摘要汇总数据,在大脑 - 小脑中执行共定位分析:
gwasDF <- fread("http://bioinfo.szbl.ac.cn/xQTL_biolinks/xqtl_data/gwasDFsub.txt")
tissueSiteDetail="Brain - Cerebellum"
head(gwasDF)
输入的数据必须要有以下几列,列名可以不一样,但顺序必须相同:
-
列1.
variants,使用rsID(例如“rs11966562”) -
列2.
chromosome,染色体 -
列3.
position,snp的基因组位置 -
列4.
P-value -
列5.
MAF,等位基因频率 -
列6.
beta,效应大小 -
列7.
se,标准误
示例使用的是内置的 GTEX 数据,可以使用以下代码查看其中可使用的组织来源 eqtl 数据:
xQTLbiolinks::tissueSiteDetailGTExv8$tissueSiteDetail %>% unique()
2.鉴定哨兵 snps
哨兵 SNP 可以通过使用 xQTLanalyze_getSentinelSnp 及其参数 p-value < 5e-8 和 SNP-to-SNP distance > 10e6 bp来检测。如果提供的 GWAS 文件基因组版本是 GRCh37,推荐将其转换为 GRCh38(使用参数:genomeVersion="grch37)。
sentinelSnpDF <- xQTLanalyze_getSentinelSnp(gwasDF, pValueThreshold = 5e-08)
# 筛选过后的snp如下
sentinelSnpDF
#> rsid chr position pValue maf beta se
#> 1: rs13120565 chr4 10702513 5.66e-10 0.6429 0.01825 0.002943.确定 snps 相关性状基因
性状基因是指位于哨兵 SNPs 1Mb 范围内(默认值,可以通过参数detectRange更改)的基因,使用函数 xQTLanalyze_getTraits 搜索哨兵 SNPs 1Mb 范围内的每个基因。
traitsAll <- xQTLanalyze_getTraits(sentinelSnpDF, detectRange=1e6, tissueSiteDetail=tissueSiteDetail)
# 总共检测到3个特征基因与1个SNP之间的3个关联
traitsAll
#> rsid chr position pValue maf beta se gencodeId
#> 1: rs13120565 chr4 10702513 5.66e-10 0.6429 0.01825 0.00294 ENSG00000002587
#> 2: rs13120565 chr4 10702513 5.66e-10 0.6429 0.01825 0.00294 ENSG00000109684
#> 3: rs13120565 chr4 10702513 5.66e-10 0.6429 0.01825 0.00294 ENSG000002614904.进行共定位分析
共定位方法中有四个统计假设:
-
H0:表型1(GWAS)和 表型2 (*QTL)与某个基因组区域的所有SNP位点无显著相关
-
H1/H2:表型1(GWAS)或表型2(*QTL)与某个基因组区域的SNP位点显著相关
-
H3:表型1(GWAS)和 表型2 (*QTL)与某个基因组区域的SNP位点显著相关,但由不同的因果变异位点驱动
-
H4:表型1(GWAS)和 表型2 (*QTL)与某个基因组区域的SNP位点显著相关,且由同一个因果变异位点驱动
① 对于单个特征基因,例如上步骤中的 ENSG00000109684,可以使用coloc方法执行共定位分析:
output <- xQTLanalyze_coloc(gwasDF, "ENSG00000109684", tissueSiteDetail=tissueSiteDetail) # using gene symbol
# 输出是一个列表,包括两部分:coloc_Out_summary 和 gwasEqtlInfo
output$coloc_Out_summary
#> nsnps PP.H0.abf PP.H1.abf PP.H2.abf PP.H3.abf PP.H4.abf
#> 1: 7107 7.097893e-11 1.32221e-07 3.890211e-06 0.00625302 0.993743
#> traitGene candidate_snp SNP.PP.H4
#> 1: ENSG00000109684 rs13120565 0.5328849
② 对于多个特征基因,可以使用for循环或lapply函数来获取所有基因的输出(使用coloc和hyprcoloc方法)
outputs <- rbindlist(lapply( unique(traitsAll$gencodeId), function(x){ # using gencode ID.
xQTLanalyze_coloc(gwasDF, x, tissueSiteDetail=tissueSiteDetail, method = "Both")$colocOut }))
# outputs是一个data.table,它合并了所有基因的 coloc_Out_summary 的所有结果
outputs
#> traitGene nsnps PP.H0.abf PP.H1.abf PP.H2.abf PP.H3.abf
#> 1: ENSG00000002587 6452 1.730175e-05 3.218430e-02 6.603361e-05 0.12198838
#> 2: ENSG00000109684 7107 7.097893e-11 1.322210e-07 3.890211e-06 0.00625302
#> 3: ENSG00000261490 6601 5.287051e-05 9.848309e-02 4.801374e-04 0.89435622
#> PP.H4.abf candidate_snp SNP.PP.H4 hypr_posterior hypr_regional_prob
#> 1: 0.84574398 rs13120565 0.4140146 0.5685 0.9694
#> 2: 0.99374296 rs13120565 0.5328849 0.9793 0.9999
#> 3: 0.00662768 rs13120565 0.4219650 0.0000 0.0101
#> hypr_candidate_snp hypr_posterior_explainedBySnp
#> 1: rs13120565 0.2726
#> 2: rs13120565 0.4747
#> 3: rs13120565 0.41185. 结果可视化
对于潜在的因果基因ENSG00000109684(PP4=0.9937 & hypr_posterior=0.9999,数值越大越显著),我们可以获得其跨组织的显著关联:
xQTLvisual_eqtl("ENSG00000109684")
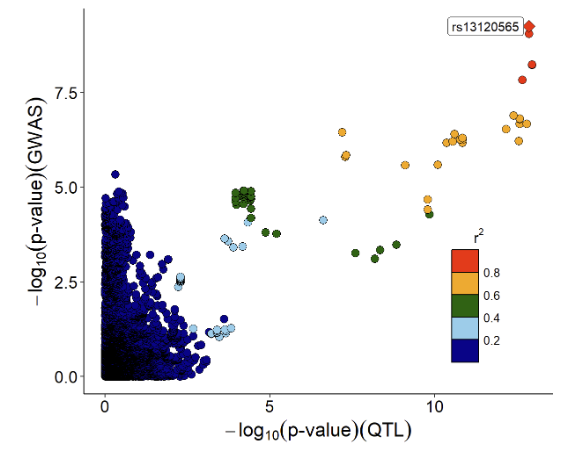
为了可视化 p 值分布和比较 GWAS 和 eQTL 的信号,首先通过 rsid 合并 GWAS 和 eQTL 的变异:
eqtlAsso <- xQTLdownload_eqtlAllAsso(gene="ENSG00000109684",
tissueLabel = tissueSiteDetail, data_source = "liLab")
gwasEqtldata <- merge(gwasDF, eqtlAsso, by="rsid", suffixes = c(".gwas",".eqtl"))
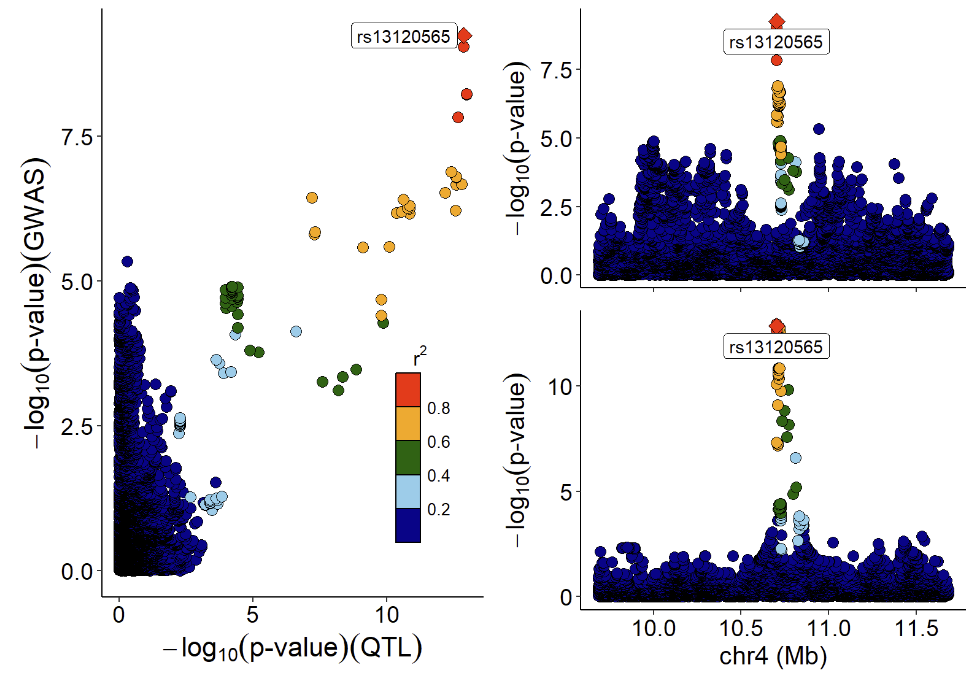
函数 xQTLvisual_locusCompare 在右上角显示候选 SNP rs13120565:
xQTLvisual_locusCompare(gwasEqtldata[,.(rsid, pValue.eqtl)],
gwasEqtldata[,.(rsid, pValue.gwas)], legend_position = "bottomright")
# 注意:SNP连锁不平衡信息会自动在线下载。由于网络问题,下载有时可能会失败。如果这种情况发生,请再次尝试运行。
GWAS 信号的 Locuszoom 图:
xQTLvisual_locusZoom(gwasEqtldata[,.(rsid, chrom, position, pValue.gwas)], legend=FALSE)
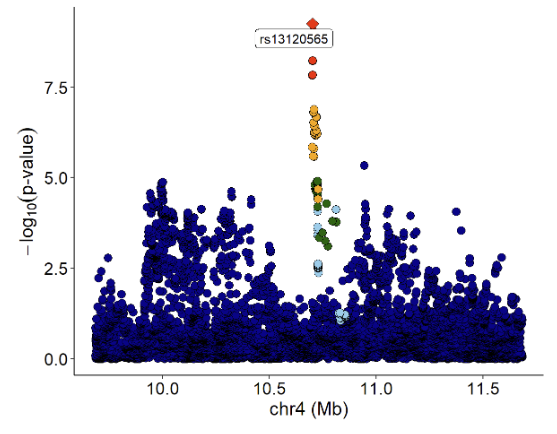
eQTL 信号的 LocusZoom 图:
xQTLvisual_locusZoom(gwasEqtldata[,.(rsid, chrom, position, pValue.eqtl)],
highlightSnp = "rs13120565", legend=FALSE)
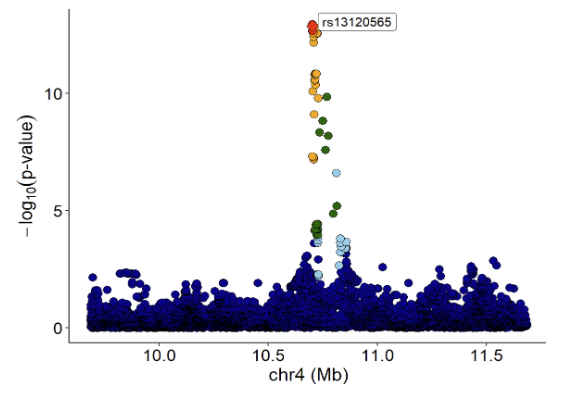
eQTL(rs13120565-ENSG00000187323.11)标准化表达的小提琴图:
xQTLvisual_eqtlExp("rs13120565", "ENSG00000109684", tissueSiteDetail = tissueSiteDetail)
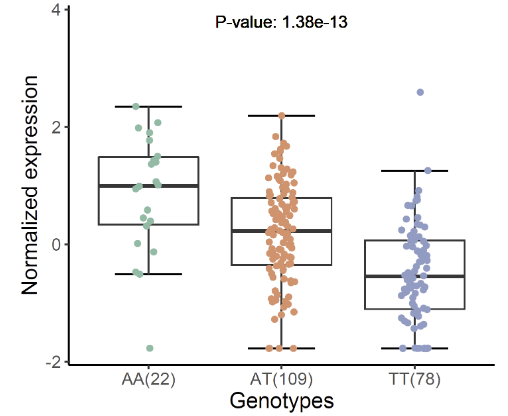
xQTLvisual_locusCombine结合locuscompare和locuszoom图:
xQTLvisual_locusCombine(gwasEqtldata[,c("rsid","chrom", "position", "pValue.gwas", "pValue.eqtl")],
highlightSnp="rs13120565")
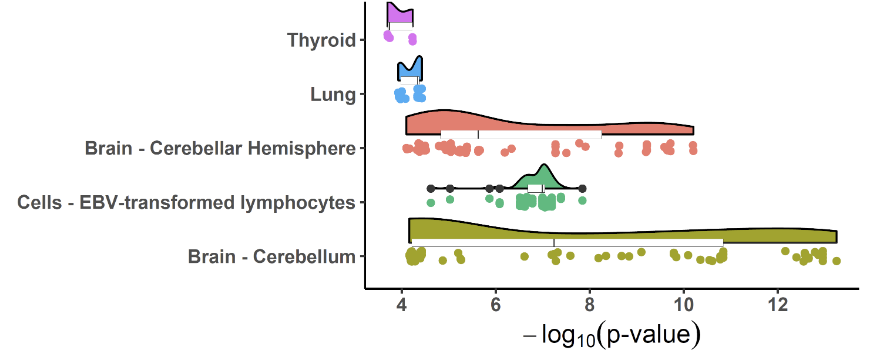
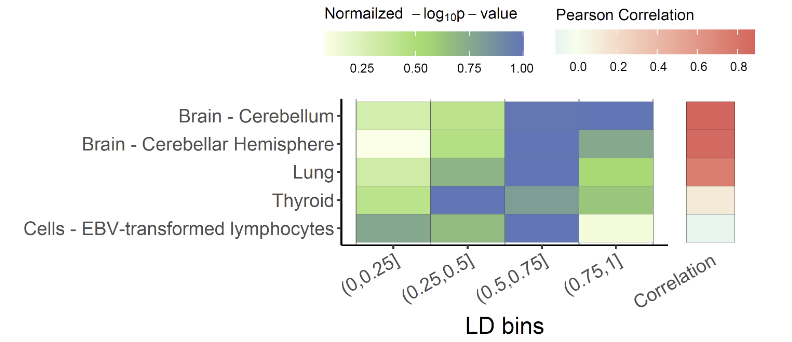
共定位位点应显示出一般模式,其中高 LD 的 SNPs 将显示出与共定位基因表达水平的强关联,而低 LD 的 SNPs 的 eQTL 关联将减弱。eQTL 的这种模式在不同组织/细胞类型中变化,其强度表明了变异的调节效应的力量。我们可以使用 xQTLvisual_coloc 可视化不同组织/细胞类型中 eQTL 的 p 值与 LD 之间的相关性,以轻松区分这种模式:
multi_tissue_coloc <- xQTLvisual_coloc(gene="ENSG00000109684", variantName="rs13120565",
tissueLabels = c("Brain - Cerebellar Hemisphere",
"Brain - Cerebellum", "Thyroid", "Lung",
"Cells - EBV-transformed lymphocytes"))
速来速去

今天就分享到这



















 2944
2944

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











