







(1)GWAS Catalog
关于GWAS Catalog数据库的下载,本文主要使用gwasrappid实现。
#使用gwasrappid
library(gwasrapidd)
help(package="gwasrappid")
#获取研究
my_studies<-get_studies(study_id = 'GCST000858', variant_id = 'rs12752552')
#示例1:寻找与自身免疫相关的靶点
my_studies <- get_studies(efo_trait = 'autoimmune disease')
gwasrapidd::n(my_studies)#显示获得了多少项研究
my_studies@studies$study_id#获取研究ID
my_studies@publications$title#研究标题
open_in_pubmed(my_studies@publications$pubmed_id)#打开浏览器
my_associations <- get_associations(study_id = my_studies@studies$study_id)#
gwasrapidd::n(my_associations)#得到182个关联
dplyr::filter(my_associations@associations, pvalue < 1e-6) %>% # 根据P值筛选
tidyr::drop_na(pvalue) %>%
dplyr::pull(association_id) -> association_ids # 提取关联ID
my_associations2 <- my_associations[association_ids]
gwasrapidd::n(my_associations2)#180个
#展示变体、风险等位基因和风险频率
my_associations2@risk_alleles[c('variant_id', 'risk_allele', 'risk_frequency')](2)UK Biobank
http://www.nealelab.is/uk-biobank

使用wget下载:如何在windows中安装wget,参考:wget 的安装与使用(Windows)_windows wget_Billie使劲学的博客-CSDN博客
win+R后再输入框内输入cmd,打开命令端后,输入下载链接。这个xlsx表格可在本文的资源绑定处下载。
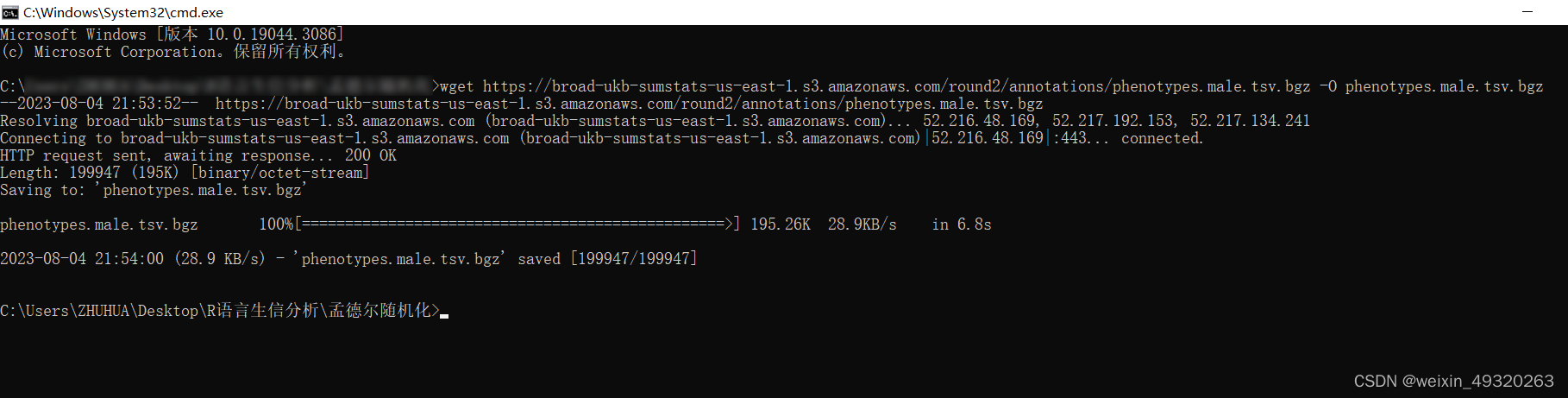
在目录中即可查看到下载的文件。接下来需要整理数据。
library(data.table)
snp150_hg19=fread('snp150_hg19.txt',data.table=F)
save(snp150_hg19,file ='snp150_hg19.Rdata')
load('snp150_hg19.Rdata')
#读取下载的GWAS数据
GWAS_1=fread('100001_irnt.gwas.imputed_v3.female.tsv',data.table = F)
head(GWAS_1)
GWAS_1$`chromosome:start`=paste0(GWAS_1$chr,':',GWAS_1$pos)
GWAS_1 <- GWAS_1[,c(11,1:10)]
GWAS_1[1:10,]
data=dplyr::inner_join(GWAS_1,snp150_hg19,by='chromosome:start')
head(data)
save(data,file ='100001_irnt_gwas_imputed_v3_female.Rdata')snp150_hg19.txt的下载链接:
链接:https://pan.baidu.com/s/1sAug3RugJy7eXAUHZhi7rA?pwd=1234
提取码:1234


















 2336
2336

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









