







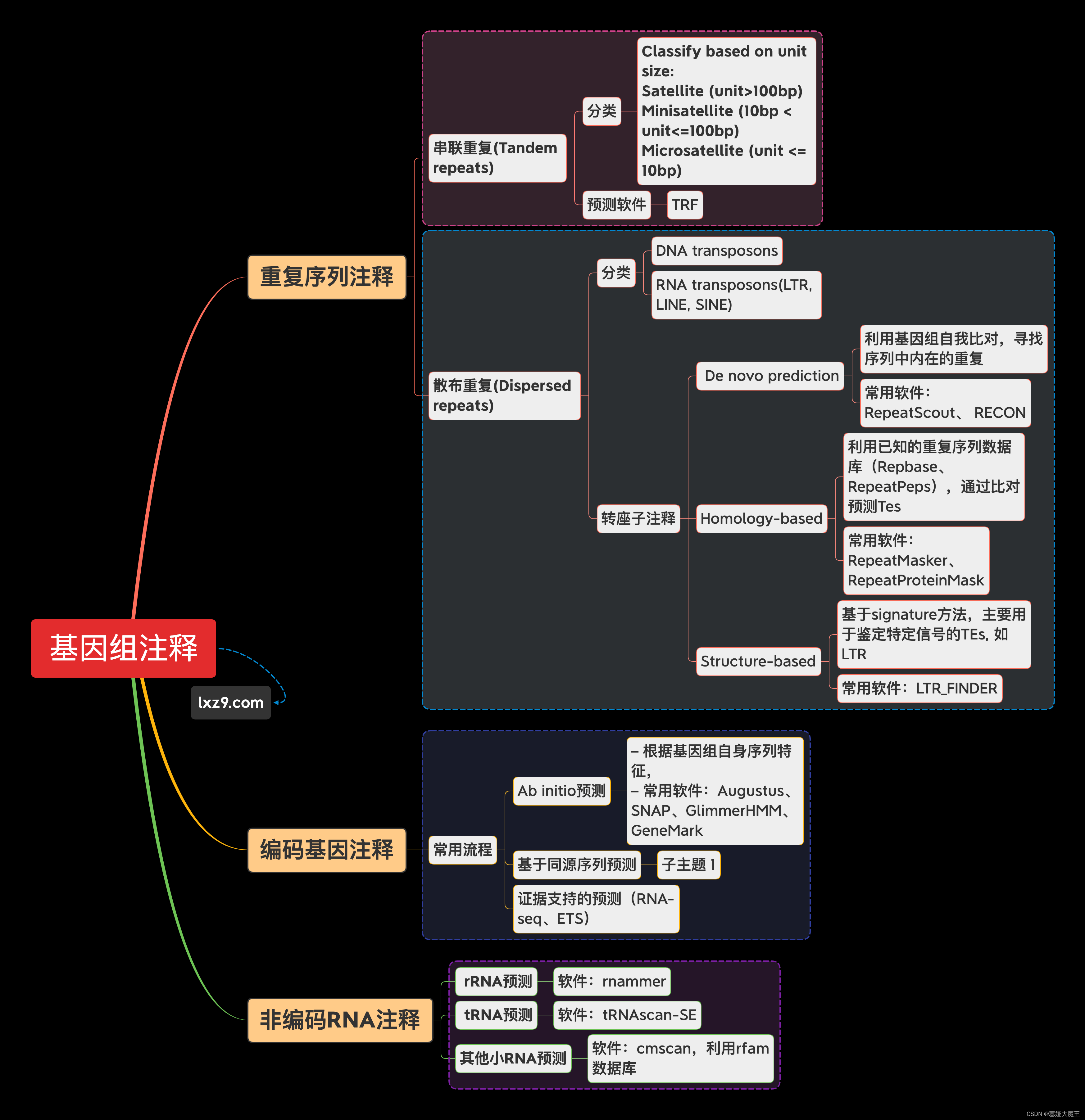
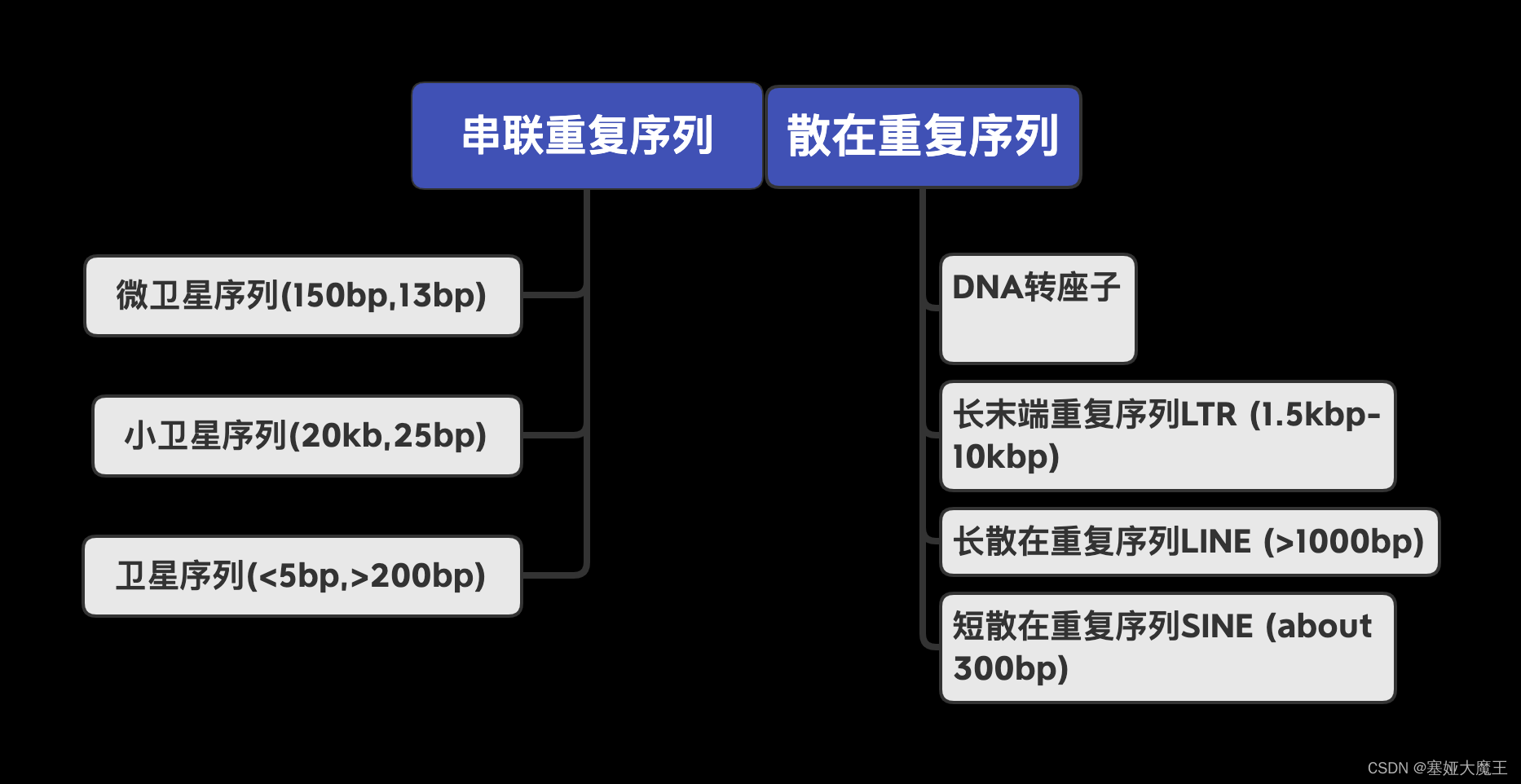
名词讲解:转座子(TE)、LTR、假基因 - 简书转座子(TE,散在重复)特征及其对基因组影响 - 简书 (jianshu.com)名词讲解:转座子(TE)、LTR、假基因 - 简书
如何进行基因组注释_genscan程序_生信技术的博客-CSDN博客
基因组注释基本流程 - Science and Technology Notes
科学网—基因组重复序列之标准分析(de novo重复序列库) - 贾安强的博文TE的鉴定 - 简书 (jianshu.com)科学网—基因组重复序列之标准分析(de novo重复序列库) - 贾安强的博文
转座子的鉴定方法基本归于两大类:从头预测、基于同源比对。
不同的算法实现的目标有所不同, 如从头算法主要是为了发现新的转座子, 因此常用来在新测序的基因组中鉴别新的转座子,而多数基于同源性算法的软件主要被用来注释基因组中的转座子。
从头预测算法 de novo 包括:基于基因组序列比对的方法、K-mer 方法、基于结构特征的方法
从头算法鉴别转座子的原理是基于转座子的重复特征,该算法可在不借助任何转座子数据库的情况下查找到几乎所有类型的转座子。
优点:算法主要用于发现新的、未注释的转座子家族, 对于高频出现的转座子鉴别尤其有效。
缺点:计算量大。另外, 由于从头算法是基于用一个转座子的拷贝数来定义重复家族, 这样低拷贝数的转座子可能被忽略掉。
基于基因组序列比对的方法。该方法利用 BLAST 等软件将基因组与基因组进行比对, 然后将双序列比对的结果转换成多序列比对, 最后用聚类方法将相关序列聚成家族,从而得到重复序列( 包括转座子) 家族。代表:RECON
K-mer 方法。 该类从头算法检索重复出现的定长 Kmer 种子( 序列短串) , 然后再将它扩展为更长的序列。代表:RepeatScout。软件首先在未知的基因组序列中计算出所有定长 K-mer 种子出现的频率, 再选择出最高频率的 K-mer 种子及其周围区域的序列, 一次一个碱基向两边扩展, 每次生成一条具有代表性 K-mer 重复家族的共有序列。然后调整已出现过的 K-mer 频率数, 再选择出包括调整过的最高频率 K-mer 及其周围区域序列, 扩展并产生共有序列,直到最高频率到达所设定的最小阈值结束,这样就得到了这一基因组的转座子家族。RepeatScout与 RECON 相比, 所得到结果更加准确,而且敏感度和运行速度都有很大的提高。其他使用 K-mer 方法的软件还有RepeatFinder等。
基于结构特征的算法。 转座子中 LTR 逆转录转座子、SINE、微型反向重复转座元件( MITE) 、Helitron 等都具有较明显的特征, 基于结构特征的算法可根据这些特征对这些转座子进行鉴别。代表:LTR_STRUC,LTR_FINDER,LTRharvest ,LTRdigest。LTR_finder和 LTRharvest是目前为止鉴定 LTR 最敏感的程序,但假阳性依然很高。
① RepeatModeler,LTR_retriever
RepeatModeler 利用全基因组序列从头预测(de novo),训练重复序列集构建本地 repeat library。RepeatModeler1.0 核心组件是 RECON(de novo,基于基因组序列比对)和RepatScout(de novo,基于 K-mer)。RepeatModeler2.0更新后加入了 LtrHarvest(de novo,基于结构),LTR_retriever(主要有LTR_FINDER,LTRharvest,都是 de novo 基于结构) 等,可以识别LTR的结构。
公司的重复序列注释流程:使用RepeatModeler从头鉴定(都是 de novo,1.0基于基因组序列比对,基于 K-mer;2.0 基于基因组序列比对,基于 K-mer,基于结构)重复区域家族,生成repeat library。然后再用RepeatMasker(基于同源性)鉴别基因组上的重复区域。
②用LTRharvest和LTRdigest进行LTR基于结构的从头预测(de novo,基于结构)
2017 年密歇根州立大学园艺系的 Shujun Ou 团队开发LTR_retriever平台用于LTR的鉴定,文章发表在Plant Physiology上。这是一款整合软件,以一或多个 LTR 预测软件鉴定 LTR 的结果作为输入文件,通过不同模块对 LTR 进行过滤和修正来对预测软件的预测结果进行整合和调整,得到非冗余精准且完整的物种特异 LTR 库,再使用RepeatMasker进行预测。
LTR_retriever不是一个独立的工具,他的主要作用就是整合 LTRharvest, LTR_FINDER, MGEScan 3.0.0, LTR_STRUC, 和 LtrDetector的结果,过滤其中的假阳性LTR-RT,得到高质量的LTR-RT库。尽管LTR_retriever支持多个LTR工具的输入,但其实上LTRharverst和LTR_FINDER的结果就已经很不错了。目前推荐的是LTR_Finder(de novo,基于结构)和LTR_harvest(de novo,基于结构)组合鉴定,之后使用LTR_retreiver整合两者的结果。
基于同源性的方法包括:基于同源序列比对的方法、基于隐马尔柯夫模型( HMM) 的方法
基于同源性的算法是将一条未知序列与已知的转座子序列或序列特征模型进行比较,从而鉴别转座子的一类方法。基于同源序列比对的算法。该类算法与从头算法中的基于基因组序列比对的方法都是使用 BLAST 等工具来发现序列相似性, 但与后者不同的是, 基于同源序列比对的方法是将未知序列与数据库中的转座子序列进行比较来鉴别转座子。转座子数据库可使用公共数据库 Repbase,但在自己物种的研究,基本都是通过当前的全基因组序列,训练重复序列集构建本地repeat library,再通过RepeatMasker注释重复序列。其中,与RepeatMasker配套的RepeatModeler,可以实现。代表:RepeatMasker。RepeatMasker 利用 BLAST 工具在转座子数据库(Repbase或者自己构建的repeat library)中比对查找已知的重复因子家族, 是目前基因组转座子注释最常用的软件。
参考文献如下
JoF | Free Full-Text | Phylogenomics and Comparative Genomics Highlight Specific Genetic Features in Ganoderma SpeciesReconstructing the evolutionary history of gypsy retrotransposons in the Périgord black truffle (Tuber melanosporum Vittad.) | SpringerLinkTransposable elements (TEs) were identified as described in Payen et al. [56]. Briefly, de novo repeat sequences were predicted in unmasked genome assemblies of 58 genomes, using RepeatScout 1.0.6 [57]. Sequences ≥ 100 bp and ≥10 occurrences were filtered out. The selected sequences were annotated by searching homologous sequences against the fungal references in REPBASE v.22.08 (Current release of Repbase Update - GIRI, accessed on 16 January 2022) using tBLASTx [58]. The coverage of TEs in the genomes, including unknown TEs, was estimated by REPEATMASKER open 4.1.1 (RepeatMasker Home Page, accessed on 16 January 2022).
RepeatScout (Price et al. 2005) was used to identify de novo repetitive DNA sequences in the T. melanosporum Mel28 genome version 1.0 (Martin et al. 2010), which is available at the Institut National de la Recherche Agronomique (INRA) Tuber genome database (http://mycor.nancy.inra.fr/IMGC/TuberGenome/download.php?select=fast).
The default parameters (with l = 15) were used. RepeatScout generated a library of consensus sequences of repeated sequences identified in the genome.
The RepeatScout library was then filtered as follows:
(1) all the sequences with a size less than 100 bp were discarded;
(2) repeats with less than ten copies in the genome were removed (because they may correspond to protein-coding gene families);
(3) repeats with significant hits to known proteins in UNIPROT (http://www.uniprot.org), other than proteins known as belonging to TEs, were removed.
The consensus sequences remaining in the RepeatScout library corresponding to repeated sequences were manually annotated by a TBLASTX search (Altschul et al. 1990) against REPBASE (version 16.03; Jurka et al. 2005). The 271 putative full-length gypsy retrotransposons identified by Martin et al. (2010) using LTR_STRUC (McCarthy and McDonald 2003) were also included in this study. The number of occurrences and the percent of genome coverage were assessed by masking the genome assemblies using REPEATMASKER version open-4.0.5 (http://www.repeatmasker.org) with the consensus sequences coming from the RepeatScout and LTR_STRUC pipelines.
简而言之,使用 RepateScout,在未掩盖的58个基因组的基因组组装中预测了从头重复序列。筛选出≥100bp 和≥10个出现的序列。通过使用 tBLASTx 搜索 REPBASE 中真针对菌参考的同源序列来注释所选序列。REPEATMASKER估计了基因组中 TE 的覆盖率,包括未知的 TE。
使用RepeatScout (Price et al. 2005)鉴定从头重复的 DNA 序列。使用了缺省参数(l = 15)。RepateScout 生成了一个基因组中鉴定的重复序列的共有序列文库。
(1)丢弃所有大小小于100bp 的序列;
(2)去除基因组中少于10个拷贝的重复序列(因为它们可能对应于蛋白质编码基因家族) ;
(3)去除对 UNIPROT 中已知蛋白质具有显着命中的重复序列( http://www.UNIPROT.org ) ,属于 TE 的蛋白质除外。
通过针对 REPBASE (版本16.03; Jurka 等,2005)的 TBLASTX 搜索(Altschul 等,1990)手动注释与重复序列相对应的 RepeatScout 文库中的共有序列。Martin 等人(2010)使用 LTR _ STRUC (McCarthy 和 McDonald 2003)鉴定的271个推定的全长吉普赛逆转录转座子也包括在本研究中。通过使用 REPEATMASKER 版本 open-4.0.5( http://www.REPEATMASKER.org )掩盖基因组组装,并使用来自 repeat atScout 和 LTR _ STRUC 管道的共有序列来评估出现的次数和基因组覆盖率。在 INRA Tuber 基因组数据库( http://mycor.nancy.INRA.fr/imgc/tubergenome/download.php?select=anno )中,可以找到 repeat atScout 共有序列和推定的全长吉普赛元素。
repeatscount | 生信菜鸟团
GitHub - mmcco/RepeatScout: The RepeatScout 1.0.5, written by Pevzner et al., source code for browsing. The official release and more information are available at http://bix.ucsd.edu/repeatscout/
方法一:RepeatScout
第一步:用build_lmer_table命令把整个基因组生成一个频率表格,把所有有过重复的kmer都找出来。
build_lmer_table
# 参数如下
build_lmer_table Version 1.0.6
Usage:
build_lmer_table -l <l> -sequence <seq> -freq <output> [opts]
-tandem <d> --- tandem distance window (def: 500)
-min <#> --- smallest number of required lmers (def: 3)
-v --- output a small amount of debugging information.
运行
build_lmer_table -l 15 -sequence /media/aa/DATA/SZQ2/bj/my/genome/5.1210/04genome_feature_analysis_primary/repeat_analysis/genome.fasta -freq 5.1210.freq第二步:用 RepeatScout 这个命令根据生成的频率表格和基因组序列产生一个包含有所有的能找到的重复元件的文件。
# 进入环境
sudo su
密码
conda activate training
# 查看参数
RepeatScout -h
# 如下
RepeatScout Version 1.0.6
Usage:
RepeatScout -sequence <seq> -output <out> -freq <freq> -l <l> [opts]
-L # size of region to extend left or right (10000)
-match # reward for a match (+1)
-mismatch # penalty for a mismatch (-1)
-gap # penalty for a gap (-5)
-maxgap # maximum number of gaps allowed (5)
-maxoccurrences # cap on the number of sequences to align (10,000)
-maxrepeats # stop work after reporting this number of repeats (10000)
-cappenalty # cap on penalty for exiting alignment of a sequence (-20)
-tandemdist # of bases that must intervene between two l-mers for both to be counted (500)
-minthresh # stop if fewer than this number of l-mers are found in the seeding phase (3)
-minimprovement # amount that a the alignment needs to improve each step to be considered progress (3)
-stopafter # stop the alignment after this number of no-progress columns (100)
-goodlength # minimum required length for a sequence to be reported (50)
-maxentropy # entropy (complexity) threshold for an l-mer to be considered (-.7)
-v[v[v[v]]] How verbose do you want it to be? -vvvv is super-verbose.
运行
# -goodlength 100 丢弃所有大小小于100bp 的序列
RepeatScout -sequence /media/aa/DATA/SZQ2/bj/my/genome/5.1210/04genome_feature_analysis_primary/repeat_analysis/genome.fasta -freq 5.1210.freq -l 15 -output 5.1210_repeat -goodlength 100Third, the "filter-stage-1.prl" script is run on the output of RepeatScout to remove low-complexity and tandem elements; RepeatMasker is run on the sequence of interest using this filtered RepeatScout library.
# 示例
cat repeats.fa | filter-stage-1.prl > repeats-filtered.prl
# 运行
cat 5.1210_repeat | filter-stage-1.prl > repeats-filtered.prl
# RepeatMasker
mkdir Repeat_result
RepeatMasker -e ncbi -lib repeats-filtered.prl -pa 16 -dir Repeat_result -gff /media/aa/DATA/SZQ2/bj/my/genome/5.1210/04genome_feature_analysis_primary/repeat_analysis/genome.fastaThe program "filter-stage-2.prl" then filters out any repeat element that does not appear a certain number of times (by default, 10). 去除基因组中少于10个拷贝的重复序列 Note that this requires you to have run RepeatMasker on a sequence with the library.
# 示例
cat repeats.lib | ./filter-stage-2.prl --cat=repeats.out --thresh=20
# repeats.out:The RepeatMasker output file. It can either be a .cat file or a .out file, but a .out file is preferred.
# 运行
cat repeats-filtered.prl | filter-stage-2.prl --cat=/media/aa/DATA/SZQ2/bj/my/genome/5.1210/04genome_feature_analysis_primary/repeat_analysis/RepeatScout/Repeat_result/genome.fasta.out --thresh=10 &> repeats-filtered.pr2Finally, the locations of the repeats found by RepeatMasker are used, in conjuction with GFF files that describe segmental duplications or exons or other such "uninteresting" regions to remove sequences from the library that are likely to not be mobile elements; the program "compare-out-to-gff.prl" does exactly this.
# RepeatMasker
mkdir Repeat_result2
RepeatMasker -e ncbi -lib repeats-filtered.pr2
-pa 16 -dir Repeat_result2 -gff /media/aa/DATA/SZQ2/bj/my/genome/5.1210/04genome_feature_analysis_primary/repeat_analysis/genome.fasta
# 示例
compare-out-to-gff.prl --gff=file1.gff --gff=file2.gff --cat=repeatmasker.out --f=file.fa > lib.ref
--gff:A GFF-formatted file of features. More than one file may be specified with multiple --gff options.
--f:A fasta formatted file. If this is given, then sequences that are under (over) the overlap threshold will be in the output. This is a sequence filter.
# 运行
compare-out-to-gff.prl --thresh 10 --gff=/media/aa/DATA/SZQ2/bj/my/genome/5.1210/04genome_feature_analysis_primary/repeat_analysis/RepeatScout/Repeat_result2/genome.fasta.out.gff --cat=/media/aa/DATA/SZQ2/bj/my/genome/5.1210/04genome_feature_analysis_primary/repeat_analysis/RepeatScout/Repeat_result2/genome.fasta.out --f=/media/aa/DATA/SZQ2/bj/my/genome/5.1210/04genome_feature_analysis_primary/repeat_analysis/genome.fasta > lib.ref采用基于同源比对和从头搜索相结合的策略,识别测序菌株基因组中的重复序列。
对于转座元件(TE)的从头预测,使用具有默认参数的RepeatModeler(http://www.repeatmasker.org/RepeatModeler.html)、RepeatScout(Price et al.,2005)和LTR-Finder(Xu et al.,2007)。
为了对同源序列进行比对以鉴定组装基因组中的重复序列,使用RepeatProteinMask(http://www.repeatmasker.org/)、RepeatMasker(http://www.RepeatMasker.org)和重复基因库。具有相同类型的重复转座因子被整合,移除分数低且重叠超过其长度的80%的属于不同类型的转座因子。
RepeatMasker
通过使用 tBLASTx 搜索 REPBASE 中针对真菌参考的同源序列来注释所选序列
注释与重复序列相对应的 RepeatScout 文库中的共有序列
# 新建文件夹
mkdir repeatMaskerfungi
# 运行
RepeatMasker -pa 40 -e ncbi -species fungi -dir repeatMaskerfungi/ -gff /media/aa/DATA/SZQ2/bj/a.preparing_data/genome_unmasked_fasta_data/Amath1.genome.fastaRepeatModeler
/media/aa/DATA/JJH/software/RepeatModeler-2.0.1/RepeatModeler
# 参数如下
/media/aa/DATA/JJH/software/RepeatModeler-2.0.1/RepeatModeler - 2.0.1
NAME
RepeatModeler - Model repetitive DNA
SYNOPSIS
RepeatModeler [-options] -database <XDF Database>
DESCRIPTION
The options are:
-h(elp)
Detailed help
-database
The name of the sequence database to run an analysis on. This is the
name that was provided to the BuildDatabase script using the "-name"
option.
-pa #
Specify the number of parallel search jobs to run. RMBlast jobs will
use 4 cores each and ABBlast jobs will use a single core each. i.e.
on a machine with 12 cores and running with RMBlast you would use
-pa 3 to fully utilize the machine.
-recoverDir <Previous Output Directory>
If a run fails in the middle of processing, it may be possible
recover some results and continue where the previous run left off.
Simply supply the output directory where the results of the failed
run were saved and the program will attempt to recover and continue
the run.
-srand #
Optionally set the seed of the random number generator to a known
value before the batches are randomly selected ( using Fisher Yates
Shuffling ). This is only useful if you need to reproduce the sample
choice between runs. This should be an integer number.
-LTRStruct
Run the LTR structural discovery pipeline ( LTR_Harvest and
LTR_retreiver ) and combine results with the RepeatScout/RECON
pipeline. [optional]
-genomeSampleSizeMax #
Optionally change the maximum bp of the genome to sample in all
rounds of RECON (default=243000000).
CONFIGURATION OVERRIDES
-repeatmasker_dir <string>
The path to the installation of RepeatMasker.
-trf_prgm <string>
The full path including the name for the TRF program ( 4.0.9 or
higher )
-rmblast_dir <string>
The path to the installation of the RMBLAST sequence alignment
program.
-rscout_dir <string>
The path to the installation of the RepeatScout ( 1.0.6 or higher )
de-novo repeatfinding program.
-recon_dir <string>
The path to the installation of the RECON de-novo repeatfinding
program.
-ltr_retriever_dir <string>
The path to the installation of the LTR_Retriever structural LTR
analysis package.
-ninja_dir <string>
The path to the installation of the Ninja phylogenetic analysis
package.
-genometools_dir <string>
The path to the installation of the GenomeTools package.
-mafft_dir <string>
The path to the installation of the MAFFT multiple alignment
program.
-abblast_dir <string>
The path to the installation of the ABBLAST sequence alignment
program.
-cdhit_dir <string>
The path to the installation of the CD-Hit sequence clustering
package.
SEE ALSO
RepeatMasker, RMBlast
COPYRIGHT
Copyright 2005-2019 Institute for Systems Biology
AUTHOR
RepeatModeler:
Robert Hubley <rhubley@systemsbiology.org>
Arian Smit <asmit@systemsbiology.org>
LTR Pipeline Extensions:
Jullien Michelle Flynn <jmf422@cornell.edu>RepeatModeler+RepeatMasker的安装与使用 - 简书 (jianshu.com)
示例
# 第一步,将输入fasta文件建库
BuildDatabase -engine ncbi -name genome ./00.data/genome.fa
# 第二步,执行RepeatModeler软件
RepeatModeler -database genome -engine ncbi -pa 10 -LTRStruct >&run.out运行
BuildDatabase -name repeatdb_Amath1 -engine ncbi /media/aa/DATA/SZQ2/bj/a.preparing_data/genome_unmasked_fasta_data/Amath1.genome.fasta
/media/aa/DATA/JJH/software/RepeatModeler-2.0.1/RepeatModeler -engine ncbi -pa 10 -database repeatdb_Amath1 -LTRStruct &> repeatdb_5.1210.out 运行成功完成后,将生成两个文件,还有一个文件夹。
看一下repeatdb_5.1210-families.fa或~/RM_2742606.WedApr191330442023/consensi.fa.classified所有的ID,可以发现在repeatmodeler运行完就已经把基因组的所有序列进行了标识。
grep ">" repeatdb_5.1210-families.fa
grep ">" consensi.fa.classified下一步用到的只有repeatdb_5.1210-families.fa或~/RM_2742606.WedApr191330442023/consensi.fa.classified
grep -i Unknown repeatdb_Amath1-families.fa > Unknown.fa
# 将ath-families.fa分ModelerID.lib和Modelerunknown.lib
seqkit grep -nrp Unknown repeatdb_Amath1-families.fa > Modelerunknown.lib
seqkit grep -vnrp Unknown repeatdb_Amath1-families.fa > ModelerID.lib
# 其中Modelerunknown.lib用RepeatMasker或TEclass进一步注释,如果能够被分类则从Modelerunknown.lib,移动到ModelerID.lib
Institute of Bioinformatics WWU Münster
「基因组注释」使用RepeatModeler从头注释基因组的重复序列_徐洲更hoptop的博客-CSDN博客
使用TEclass对TE一致性序列进行分类_徐洲更hoptop的博客-CSDN博客
安装TEclass
wget http://www.compgen.uni-muenster.de/tools/teclass/download/TEclass-2.1.3.tar.gz
tar xf TEclass-2.1.3.tar.gz
cd TEclass-2.1.3
# 下载依赖的软件
sh Download_dependencies.sh
Build_ICM_model.pl* Configure.pl* TEclass.pm* libsvm.tar.gz*
Build_LVQ_sets.pl* Download_dependencies.sh* TEclassBuild.pl* lvq_pak.tar*
Build_Random_Forests.pl* Install.sh* TEclassTest.pl* testfile/
Build_SVM_models.pl* Preprocessing.pl* classifiers/ testfile.fa*
Compile_dependencies.sh* README* glimmer.tar.gz*由于代码老旧,部分内容无法自动下载,需要手动下载, 例如librf, blast. 最终要保证文件夹下有如下文件
libsvm.tar.gz: http://www.csie.ntu.edu.tw/~cjlin/libsvm/
glimmer.tar.gz: http://ccb.jhu.edu/software/glimmer/
librf.tar.gz: http://mtv.ece.ucsb.edu/benlee/librf.html
lvq_pak.tar: http://www.cis.hut.fi/research/som-research/nnrc-programs.shtml
blast.tar.gz: ftp://ftp.ncbi.nlm.nih.gov/blast/executables/legacy.NOTSUPPORTED
# blast
curl -o 'blast.tar.gz' ftp://ftp.ncbi.nlm.nih.gov/blast/executables/legacy.NOTSUPPORTED/2.2.26/blast-2.2.26-x64-linux.tar.gz
# librf
curl -o 'librf.tar.gz' https://github.com/tearshark/librf/archive/refs/tags/2.9.10.tar.gz
# 编译依赖的软件
sh Compile_dependencies.sh# 这里用TEclass进行分类
TEclassTest Modelerunknown.lib运行RepeatMasker
示例
RepeatMasker -e ncbi -lib hud-families.fa -pa 16 Lichenomphalia_hudsoniana_LH.genome.fasta 运行
mkdir repeatMasker2
RepeatMasker -pa 40 -e ncbi -lib repeatdb_Amath1-families.fa -dir repeatMasker2/ -gff /media/aa/DATA/SZQ2/bj/a.preparing_data/genome_unmasked_fasta_data/Amath1.genome.fasta
方法二:EDTA
使用EDTA进行TE注释_徐洲更hoptop的博客-CSDN博客
EDTA 最简易安装方法_tesorter_Plaichat-123的博客-CSDN博客repeat注释---EDTA - 简书 (jianshu.com)
EDTA | TE 注释工具 - 简书 (jianshu.com)
从它的描述中我们也可以看出它是一个综合性的流程工具,它整合了目前LTR预测工具结果,TIR预测工具结果,MITE预测工具结果,Helitrons预测工具结果, 从而构建出一高可信,非冗余的TE数据库,用做基因组的注释。所以说,不用想以前注释repeat,需要几个工具综合。EDTA(Extensive de novo TE Annotator)全称是 Extensive de-novo TE Annotator。(This package is developed for automated whole-genome de-novo TE annotation and benchmarking the annotation performance of TE libraries.)是一个可以从头注释全基因组中的TE并且评估已有TE库注释优劣的工具包。该工具的主要步骤是过滤掉原始TE中注释错误的,从而注释出全基因组中较高质量的非冗余TE库。从它的描述中我们也可以看出它是一个综合性的流程工具,它整合了目前LTR预测工具结果,TIR预测工具结果,MITE预测工具结果,Helitrons预测工具结果, 从而构建出一高可信,非冗余的TE数据库,用做基因组的注释。工具包中的一些程序的选择只是基于水稻基因组中人工校正的TE库,可能对其他基因组并不是很合适。因此,在其他基因组中使用该工具包时应该特别注意。
安装
如果没有管理员权限,可以用conda进行安装。如果有管理员权限,可以尝试用docker或者singularity进行安装。
conda
使用conda的安装方法如下
git clone https://github.com/oushujun/EDTA
cd EDTA
conda env create -f EDTA.yml
conda activate EDTA
EDTA.pl运行
# 示例(3种)
EDTA.pl -genome chr1.fa -species others -step all -t 20
EDTA.pl --genome genome.fa --cds genome.cds.fa --curatedlib ../database/rice6.9.5.liban --exclude genome.exclude.bed --overwrite 1 --sensitive 1 --anno 1 --evaluate 1 --threads 10
EDTA.pl --genome Chr1.fa --cds Chr1.cds.fa --sensitive 1 --anno 1 --evaluate 1 --threads 20 --force 1
# 参数如下
--genome: 输入的基因组序列
--species: 物种名,Rice, Maize和others三个可选
--step all: 运行步骤, all|filter|final|anno, 根据具体情况选择
--threads: 线程数,默认是4
--cds: 提供已有的CDS序列(不能包括内含子和UTR),用于过滤
--sensitive: 是否用RepeatModeler分析剩下的TE,默认是0,也就是不要。RepeatModeler运行时间比较久,量力而信。
--anno: 是否在构建TE文库后进行全基因组预测,默认是0.
--evaluate:Evaluate (1) classification consistency of the TE annotation. (--anno 1 required). Default: 0. This step is slow and does not change the annotation result.
--overwrite:如果发现以前的结果,决定是否覆盖(1,重新运行)或不覆盖(0,默认)
# 运行
mkdir EDTA && cd EDTA
EDTA.pl --genome /media/aa/DATA/SZQ2/bj/my/genome/5.1210/04genome_feature_analysis_primary/repeat_analysis/genome.fasta --cds /media/aa/DATA/SZQ2/bj/my/genome/5.1210/10.gene_prediction/evm_pasa/CDS.fasta --overwrite 1 --species others --step all --sensitive 1 --anno 1 --evaluate 1 --threads 20运行结束之后,会在当前目录下留下运行时的中间文件,保证你程序中断之后,能够断点续跑。
genome.fasta.mod.EDTA.TElib.fa是最终的TE文库(非冗余)
grep ">" genome.fasta.mod.EDTA.TElib.fa genome.fasta.mod.EDTA.TEanno.sum是repeat注释的总结文件
genome.fasta.mod.EDTA.TEanno.gff3是repeat注释的gff文件
genome.mod.EDTA.TElib.novel.fa: 新TE类型. 该文件中包括输入的修正版的TE库中没有的TE序列(需要--curatedlib参数).
genome.mod.EDTA.TEanno.gff: 全基因组TE的注释. 该文件包括结构完整和结构不完整的TE的注释(需要--anno 1参数).
genome.mod.EDTA.TEanno.sum: 对全基因组TE注释的总结(需要--anno 1参数).
genome.mod.MAKER.masked: 低阈值TE的屏蔽.该文件中仅包括长TE(>= 1 kb)序列(需要--anno 1参数).
genome.mod.EDTA.TE.fa.stat.redun.sum: 简单TE的注释偏差(需要--evaluate 1参数).
genome.mod.EDTA.TE.fa.stat.nested.sum:嵌套型TE注释的偏差(需要--evaluate 1参数).
genome.mod.EDTA.TE.fa.stat.all.sum: 注释偏差的概述(需要--evaluate 1参数)
mkdir repeatMasker
RepeatMasker -pa 40 -e ncbi -lib genome.fasta.mod.EDTA.TElib.fa -dir repeatMasker/ -gff genome.fasta最后使用方法
使用 RepeatMasker 进行基因组重复序列分析
也可使用在线网站
mkdir repeat_analysis && cd repeat_analysis
mkdir repeatMasker
# 进入conda环境
sudo su
password
conda activate training
# 运行
# pepall
for i in `cat /media/aa/DATA/SZQ2/bj/functional_annotation/99list.txt`
do
echo "RepeatMasker -pa 4 -e ncbi -species fungi -dir repeatMasker/ -gff /media/aa/Expansion/szq2/bj/a.preparing_data/genome_unmasked_fasta_data/$i.genome.fasta"
done > command.RepeatMasker.list
ParaFly -c command.RepeatMasker.list -CPU 15使用 repeatModeler 寻找本物种重复序列
mkdir repeatModeler && cd repeatModeler
# BuildDatabase
for i in `cat /media/aa/DATA/SZQ2/bj/functional_annotation/99list.txt`
do
echo "BuildDatabase -name $i -engine ncbi /media/aa/Expansion/szq2/bj/a.preparing_data/genome_unmasked_fasta_data/$i.genome.fasta"
done > command.BuildDatabase.list
ParaFly -c command.BuildDatabase.list -CPU 15
# RepeatModeler
for i in `cat /media/aa/DATA/SZQ2/bj/functional_annotation/99list.txt`
do
echo "/media/aa/DATA/JJH/software/RepeatModeler-2.0.1/RepeatModeler -engine ncbi -pa 4 -database $i -LTRStruct &> repeatdb_$i.out"
done > command.RepeatModeler.list
ParaFly -c command.RepeatModeler.list -CPU 15
# RepeatMasker
mkdir RepeatMasker2
for i in `cat /media/aa/DATA/SZQ2/bj/functional_annotation/99list.txt`
do
echo "RepeatMasker -pa 4 -e ncbi -lib /media/aa/DATA/SZQ2/bj/genome_feature/repeat_analysis/repeatModeler/$i-families.fa -dir repeatMasker2/ -gff /media/aa/Expansion/szq2/bj/a.preparing_data/genome_unmasked_fasta_data/$i.genome.fasta"
done > command.RepeatMasker2.list
ParaFly -c command.RepeatMasker2.list -CPU 15将 repeatMasker 和 repeatModeler 的统计结果合并
mkdir allstats && cd allstats
for i in `cat /media/aa/DATA/SZQ2/bj/functional_annotation/99list.txt`
do
echo "merge_repeatMasker_out.pl /media/aa/Expansion/szq2/bj/a.preparing_data/genome_unmasked_fasta_data/$i.genome.fasta ../repeatMasker/$i.genome.fasta.out ../repeatModeler/repeatMasker2/$i.genome.fasta.out > $i.genome.repeat.stats"
done > command.merge_repeatMasker_out.list
ParaFly -c command.merge_repeatMasker_out.list -CPU 15
# 根据合并后的结果,来得到 masked genome
maskedByGff.pl genome.repeat.gff3 genome.fasta --mask_type softmask > genome.softmask.fasta
maskedByGff.pl genome.repeat.gff3 genome.fasta --mask_type hardmaskX > genome.hardmaskX.fasta
maskedByGff.pl genome.repeat.gff3 genome.fasta --mask_type hardmaskN > genome.hardmaskN.fasta

















 67
67

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









