







KEGG在线数据库使用攻略_刘永鑫Adam的博客-CSDN博客
KEGG功能注释及本地化--基于KofamKOALA - 知乎
震惊!!KEGG官方工具能够完成任何物种的KEGG注释!!! - 简书
KEGG功能注释工具 KofamKOALA 安装与使用_刘永鑫Adam的博客-CSDN博客
kofamscan:基于HMM模型注释KEGG通路 再也不需要考虑数据库过时了_刘永鑫Adam的博客-CSDN博客基于HMM模型的kegg注释软件KofamKOALA - 知乎kofamscan:基于HMM模型注释KEGG通路 再也不需要考虑数据库过时了_刘永鑫Adam的博客-CSDN博客
mkdir 08.KAAS && cd 08.KAAS方法一(KAAS网页操作)
一次只能运行一个基因组!
在网页工具中提交序列进行注释。需要填写邮箱信息,在网页中提交后需要再进入邮箱,点击邮件中的提交链接,才能开始计算。
Selected organisms(up to 40 organisms):
Basidiomycetes(11):cne, cgi, tms, ppl, tvs, hir, mpr, scm, uma, mgl, mrt
Microsporidians(2):ecu, nce,
Schizosaccharomycetes(1):spo
Pezizomycetes(1):tml
Dothideomycetes(2):pno, bze
Eurotiomycetes(13):ani, afm, nfi, aor, ang, afv, pcs, pdp, cim, cpw, ure, pbl, abe
Leotiomycetes(3):ssl, bfu, psco
Sordariomycetes(7):ncr, mgr, fgr, fpu, fox, nhe, maw
Saccharomycetes(23);sce, ago, kla, kmx, lth, vpo, zro, cgr, ncs, tpf, ppa, dha, pic, pgu, spaa, lel, cal, yli, clu, clus, caur, slb, pkz汇总:
cne, cgi, tms, ppl, tvs, hir, mpr, scm, uma, mgl, mrt, ecu, nce, spo, tml, pno, bze, ani, nfi, ang, pcs, cim, ure, abe, ssl, bfu, mgr, fpu, nhe, sce, kla, lth, zro, ncs, ppa, pic, spaa, cal, clu, caur
注释完毕后,从填写的邮箱中进入注释完毕后的网页,下载注释结果query.ko文件。
如:https://www.genome.jp/tools/kaas/files/dl/1662386600/query.ko
mkdir 08.KAAS && cd 08.KAAS
cp /home/aa/jjh/服务器第二次格式化/13.functional_annotation/KEGG/ko00001.keg ko00001.keg
/media/aa/DATA/SZQ2/command/clf/bin/gene_annotation_from_kaas.pl $i.query.ko > $i.KEGG.txt
打开result目录下的html文件,将网页内容复制到.xlsx中,进行修改
方法二(eggNOG结果提取)
eggNOG官网的最终注释结果out.emapper.annotations
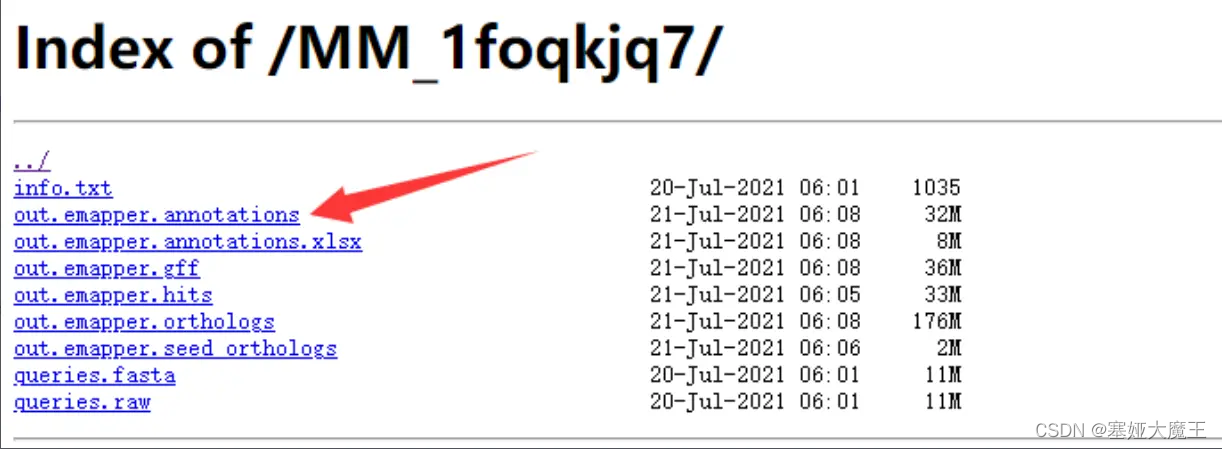
其中我们最关注的是第十列的GO号以及第十二列的KO号。
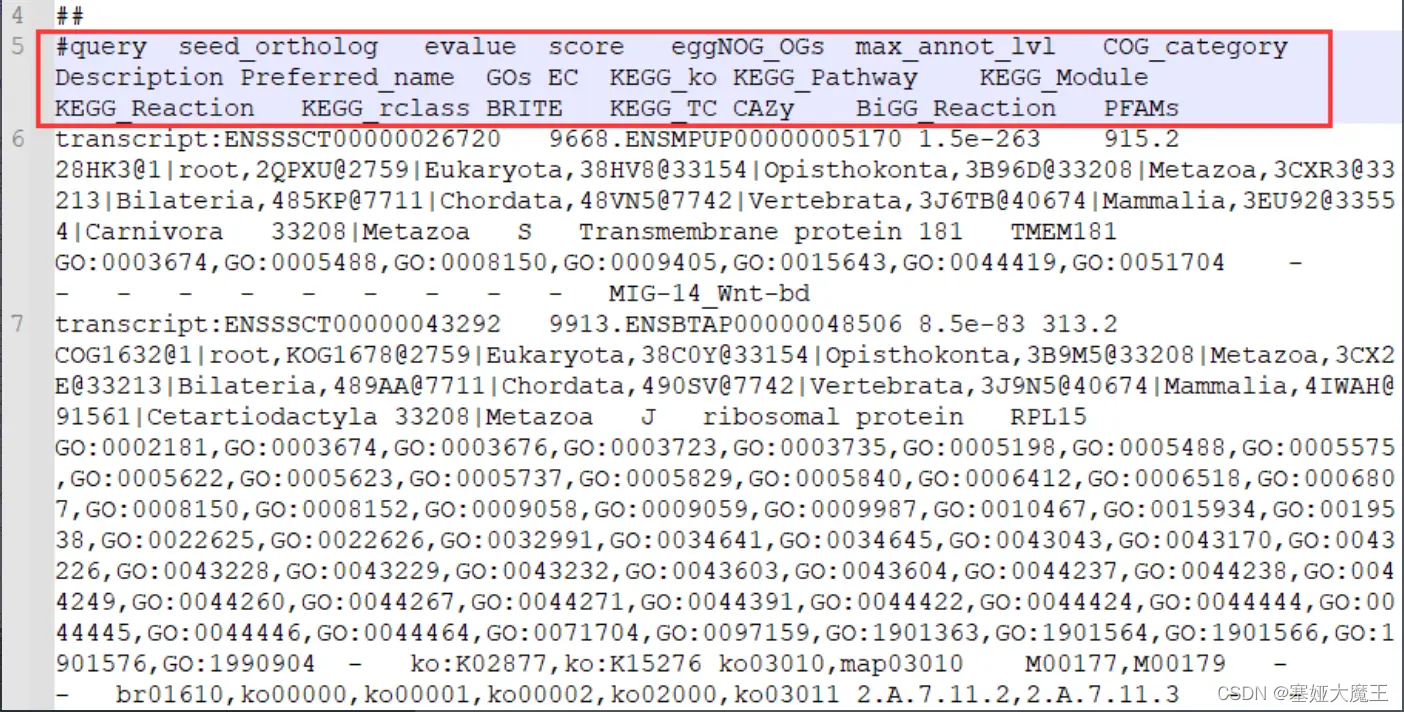
这里以提取KO号为例,制作query2ko背景文件,可以以此直接使用TBtools进行富集分析。
提取基因及其映射上的KO号,KO号在第12列。
cut -f 1,12 out.emapper.annotations > out.emapper.annotations.query2ko批量操作
批量 pep70
for i in `cat /media/aa/DATA/SZQ2/bj/functional_annotation/70list.txt`
do
echo "cut -f 1,12 /media/aa/DATA/SZQ2/bj/functional_annotation/04.eggNOG/pep70/eggnog_net70/$i.out.emapper.annotations > $i.out.emapper.annotations.query2ko"
done > command.query2ko.list
ParaFly -c command.query2ko.list -CPU 4批量 pepmy
for i in `cat /media/aa/DATA/SZQ2/bj/functional_annotation/pepmylist.txt`
do
echo "cut -f 1,12 /media/aa/DATA/SZQ2/bj/functional_annotation/04.eggNOG/pepmy/eggnog_netmy/$i.out.emapper.annotations > $i.out.emapper.annotations.query2ko"
done > command.query2ko.list
ParaFly -c command.query2ko.list -CPU 4查看提取结果发现有些基因没有被注释上,需要去掉没有注释的基因以及去掉KO号前面的
ko:
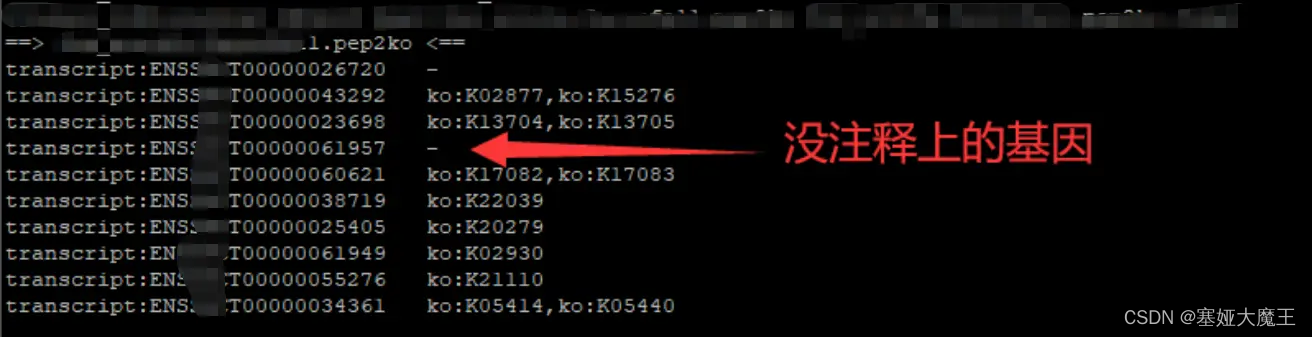
grep -v '-' out.emapper.annotations.query2ko|sed 's/ko://g' > out.emapper.annotations.query2ko.final批量操作
批量 pep70
for i in `cat /media/aa/DATA/SZQ2/bj/functional_annotation/70list.txt`
do
echo "grep -v '-' $i.out.emapper.annotations.query2ko|sed 's/ko://g' > $i.out.emapper.annotations.query2ko.final"
done > command.query2ko.final.list
ParaFly -c command.query2ko.final.list -CPU 4
批量 pepmy
for i in `cat /media/aa/DATA/SZQ2/bj/functional_annotation/pepmylist.txt`
do
echo "grep -v '-' $i.out.emapper.annotations.query2ko|sed 's/ko://g' > $i.out.emapper.annotations.query2ko.final"
done > command.query2ko.final.list
ParaFly -c command.query2ko.final.list -CPU 4整理好的query2ko文件如图
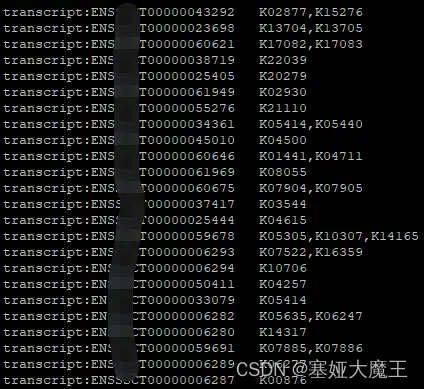
注意:需要保持ID的一致性,否则注释不出结果(例如拿来做注释的往往是蛋白序列,一般会包括可变性剪切的标识符AT4G36920.1
,这个时候需要注意做富集的差异基因是否包含该标识符,需要统一;再者,如果使用的是ensembl ID,则需要注意统一使用Transcript ID或Gene ID)。
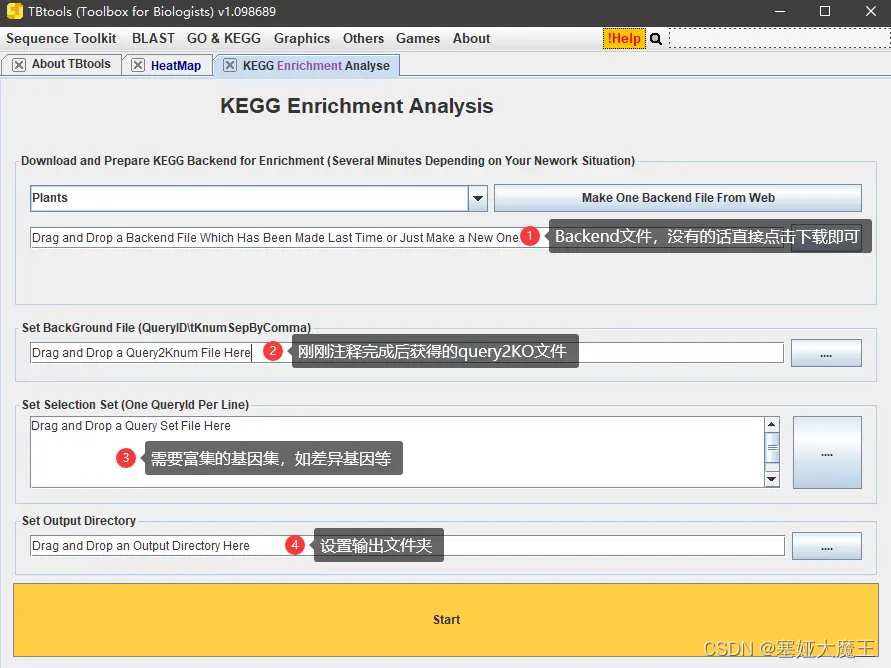
直接使用TBtools进行富集分析
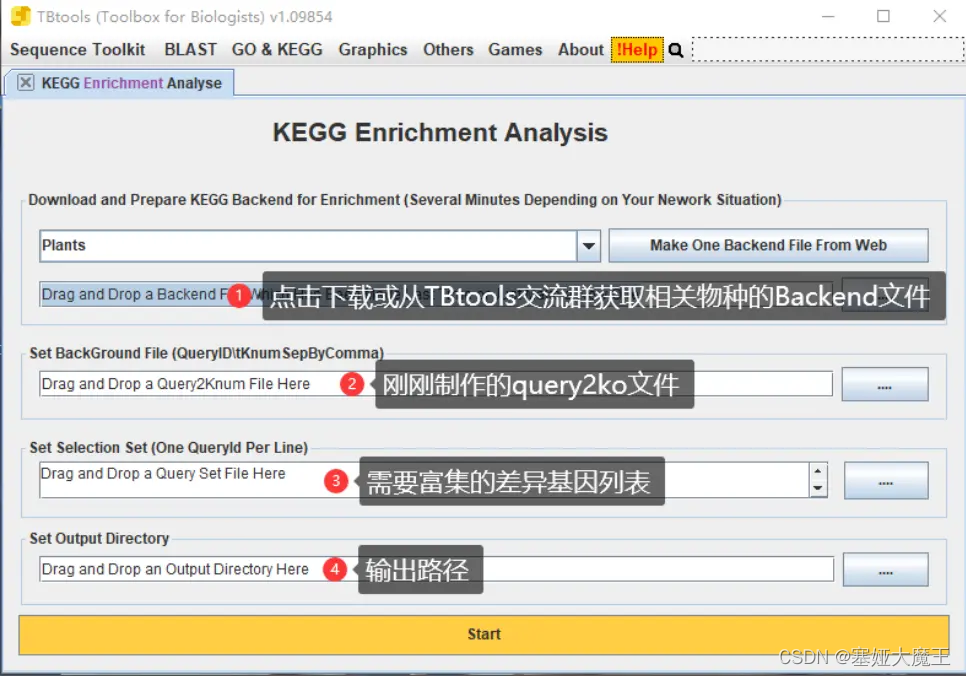
方法三(本地运行)
第1步:本地化KofamKOALA
如果需要注释的序列太多,文件太大,使用网页版工具确实可能会太慢。这个时候当然是选择将其本地化!在本地根据服务器情况提高线程加速注释。
Linux版本的KofamKOALA 需要下载KOfam(数据库)和KofamScan(软件),软件依赖Ruby,HMMER和GNU Parallel。
下载 KOfam 和 KofamScan
下载并解压KOfam
# 新建文件夹
mkdir KofamKOALA && cd KofamKOALA
# 下载
wget https://www.genome.jp/ftp/db/kofam/ko_list.gz
wget https://www.genome.jp/ftp/db/kofam/profiles.tar.gz
# 下载完成之后解压
gunzip ko_list.gz
tar -xzvf profiles.tar.gz下载并解压kofam_scan
wget https://www.genome.jp/ftp/tools/kofam_scan/kofam_scan-1.3.0.tar.gz
tar -xzvf kofam_scan-1.3.0.tar.gz将kofam_scan加入环境变量
echo export PATH=/media/aa/DATA/SZQ2/KofamKOALA/kofam_scan-1.3.0:\$PATH >> ~/.bashrc
source ~/.bashrc安装依赖,KofamScan需要Ruby,HMMER和GNU Parallel
# 版本需求
- Ruby >= 2.4
- HMMER >= 3.1
- GNU Parallel构建conda虚拟环境安装工具
conda create -n kofamscan
conda activate kofamscan
# 安装Ruby
conda install -c conda-forge ruby
# ruby-3.1.2
# 安装hmmer
conda install -c bioconda hmmer
# hmmer-3.3.2
# 安装GNU Parallel
conda install -c conda-forge parallel
# parallel-20230322-ha770c72_0 修改配置文件,指定依赖软件以及KOfam的路径
首先获取相关软件的路径(注意:配置文件中不需要指定Ruby的路径,但是需要确保Ruby加入了环境变量,能够直接调用)。
which parallel hmmscan
# 如下
/home/aa/anaconda3/envs/kofamscan/bin/parallel
/home/aa/anaconda3/envs/kofamscan/bin/hmmscan使用官方模板,修改配置文件
cd kofam_scan-1.3.0
cp config-template.yml config.yml注意,由于hmmscan和parallel都已经加入环境变量,可以直接调用,因此在配置文件中无需配置这两个软件的路径。
vi config.yml
# Path to your KO-HMM database
# A database can be a .hmm file, a .hal file or a directory in which
# .hmm files are. Omit the extension if it is .hal or .hmm file
profile: /media/aa/DATA/SZQ2/KofamKOALA/profiles
# Path to the KO list file
ko_list: /media/aa/DATA/SZQ2/KofamKOALA/ko_list
# Path to an executable file of hmmsearch
# You do not have to set this if it is in your $PATH
# hmmsearch: /home/aa/anaconda3/envs/kofamscan/bin/hmmscan
# Path to an executable file of GNU parallel
# You do not have to set this if it is in your $PATH
# parallel: /home/aa/anaconda3/envs/kofamscan/bin/parallel
# Number of hmmsearch processes to be run parallelly
cpu: 8第1步:运行
对蛋白序列进行注释(注意:注释序列必须为蛋白序列)。
exec_annotation -h
# 如下
Usage: exec_annotation [options] <query>
<query> FASTA formatted query sequence file
-o <file> File to output the result [stdout]
-p, --profile <path> Profile HMM database
-k, --ko-list <file> KO information file
--cpu <num> Number of CPU to use [1]
-c, --config <file> Config file
--tmp-dir <dir> Temporary directory [./tmp]
-E, --e-value <e_value> Largest E-value required of the hits
-T, --threshold-scale <scale>
The score thresholds will be multiplied by this value
-f, --format <format> Format of the output [detail]
detail: Detail for each hits (including hits below threshold)
detail-tsv: Tab separeted values for detail format
mapper: KEGG Mapper compatible format
mapper-one-line: Similar to mapper, but all hit KOs are listed in one line
--[no-]report-unannotated Sequence name will be shown even if no KOs are assigned
Default is true when format=mapper or mapper-all,
false when format=detail
--create-alignment Create domain annotation files for each sequence
They will be located in the tmp directory
Incompatible with -r
-r, --reannotate Skip hmmsearch
Incompatible with --create-alignment
--keep-tabular Neither create tabular.txt nor delete K number files
By default, all K number files will be combined into
a tabular.txt and delete them
--keep-output Neither create output.txt nor delete K number files
By default, all K number files will be combined into
a output.txt and delete them
Must be with --create-alignment
-h, --help Show this message and exit注意:--format参数在help中给出了四种,但主要有两种格式。
--format mapper对每一个Gene ID只保留最佳的KO Number,结果文件只含有Gene ID和KO Number的映射信息。KEGG mapper兼容的格式 (可以用于后续网页分析)
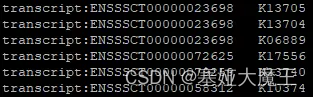
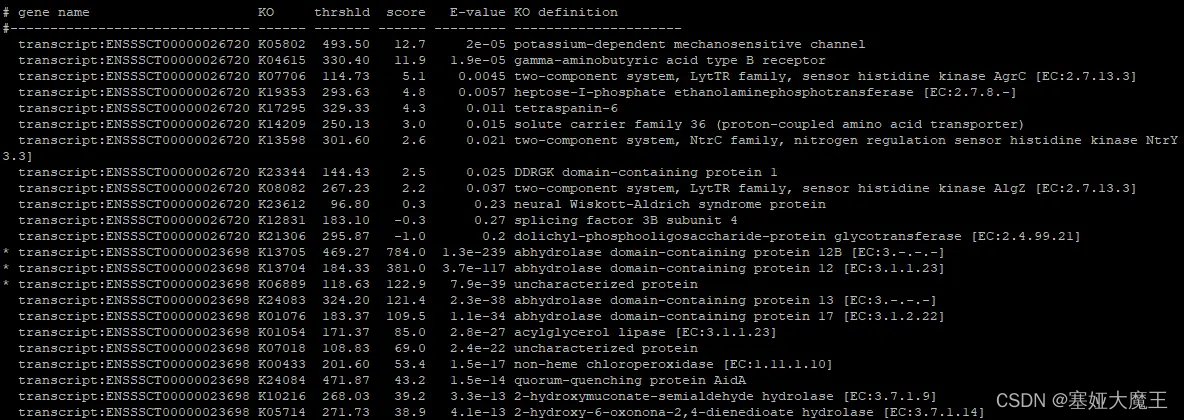
--format detail保留所有信息,包括Gene ID对应上的每一个KO Number,比对分数,E-value以及KO Number的详细信息等。
进入conda环境
conda activate kofamscanexec_annotation -o $i.querry2KO --cpu 8 --format mapper --tmp-dir $i.tmp -E 1e-5 /media/aa/DATA/SZQ2/bj/functional_annotation/pep70/$i.fasta批量操作
批量 pep70
for i in `cat /media/aa/DATA/SZQ2/bj/functional_annotation/70list.txt`
do
echo "exec_annotation -o $i.querry2KO --cpu 8 --format mapper --tmp-dir $i.tmp -E 1e-5 /media/aa/DATA/SZQ2/bj/functional_annotation/pep70/$i.fasta"
done > command.exec_annotation.list
ParaFly -c command.exec_annotation.list -CPU 48
# 或者使用detail
for i in `cat /media/aa/DATA/SZQ2/bj/functional_annotation/70list.txt`
do
echo "exec_annotation -o $i.querry2KO --cpu 8 --format detail --tmp-dir $i.tmp -E 1e-5 /media/aa/DATA/SZQ2/bj/functional_annotation/pep70/$i.fasta"
done > command.exec_annotation_detail.list
ParaFly -c command.exec_annotation_detail.list -CPU 48批量 pepmy
for i in `cat /media/aa/DATA/SZQ2/bj/functional_annotation/pepmylist.txt`
do
echo "exec_annotation -o $i.querry2KO --cpu 8 --format mapper --tmp-dir $i.tmp -E 1e-5 /media/aa/DATA/SZQ2/bj/functional_annotation/pepmy/$i.fasta"
done > command.exec_annotation.list
ParaFly -c command.exec_annotation.list -CPU 48
# 或者使用detail
for i in `cat /media/aa/DATA/SZQ2/bj/functional_annotation/pepmylist.txt`
do
echo "exec_annotation -o $i.querry2KO --cpu 8 --format detail --tmp-dir $i.tmp -E 1e-5 /media/aa/DATA/SZQ2/bj/functional_annotation/pepmy/$i.fasta"
done > command.exec_annotation_detail.list
ParaFly -c command.exec_annotation_detail.list -CPU 48最后,拿到query2KO文件之后,可以用其当背景文件,使用TBtools对基因集做富集分析。


















 4350
4350

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









