






(dauparas/ProteinMPNN: Code for the ProteinMPNN paper)
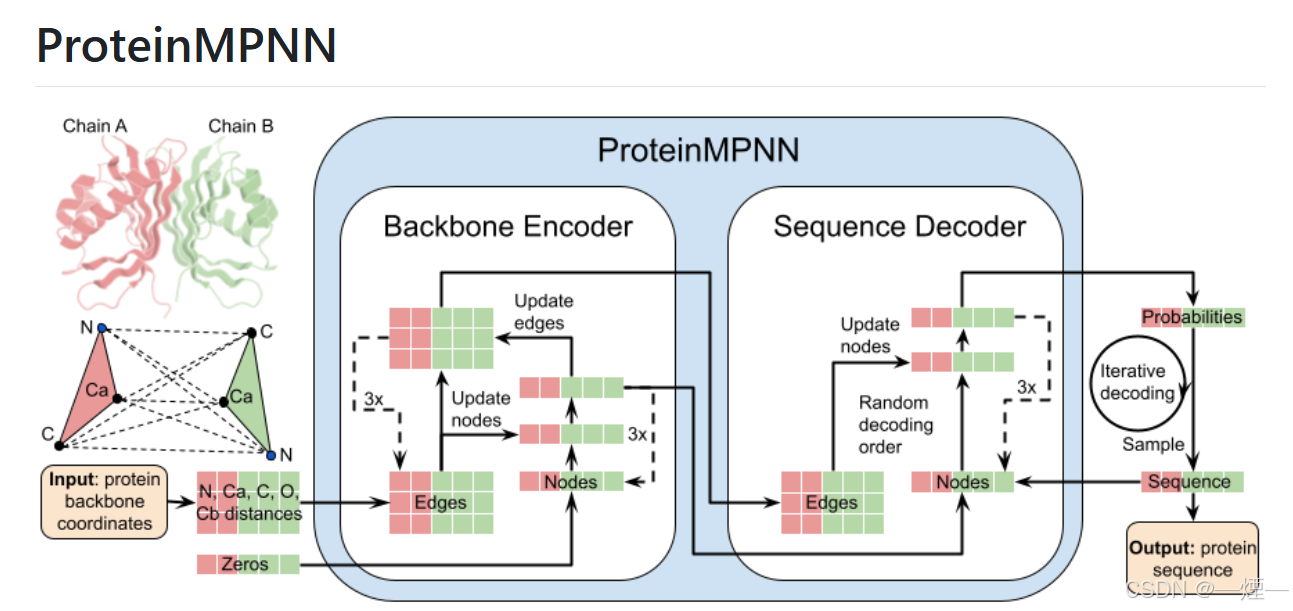
工具介绍
ProteinMPNN 是一个基于深度学习的蛋白质序列设计工具,旨在根据给定的蛋白质结构信息来设计或优化蛋白质序列。
核心功能
- 蛋白质序列设计:根据输入的蛋白质结构(PDB 文件),为其设计合适的氨基酸序列。用户可以指定设计的链、固定的残基位置、氨基酸组成偏好等条件,以满足不同的设计需求。
- 序列评分:计算给定蛋白质结构 - 序列对的得分,反映模型对该序列的不确定性。得分基于负对数概率,可用于评估设计序列的质量。
- 概率输出:可以输出条件概率和无条件概率,帮助用户了解每个位置上不同氨基酸的可能性分布。
代码组织
protein_mpnn_run.py:主脚本,用于初始化和运行模型。protein_mpnn_utils.py:包含了主脚本所需的实用函数。examples/:提供了多个简单的代码示例,涵盖了单体设计、多链设计、固定残基位置、添加氨基酸偏好等多种场景。inputs/:包含示例所需的输入 PDB 文件。outputs/:存储示例运行的输出结果。colab_notebooks/:提供了 Google Colab 示例,方便用户在云端环境中运行代码。training/:包含了重新训练模型所需的代码和数据。
安装
运行 ProteinMPNN 需要克隆 GitHub 仓库,并安装 Python(版本 >= 3.0)、PyTorch 和 Numpy。
git clone https://github.com/dauparas/ProteinMPNN.git这里我是使用conda创建了虚拟环境,使用的python 3.8
conda create --name MPNN python=3.8
激活环境
conda activate MPNN这里还安装了一个TMalign(用于计算两个蛋白质链之间的 TM 分数、RMSD 等指标)
wget https://zhanggroup.org/TM-align/TMalign.gz
gunzip TMalign.gz
chmod +x TMalign
mv TMalign /usr/local/bin/代码简介
1. StructureDataset 和 StructureLoader
- 简介:用于处理蛋白质结构数据集,
StructureDataset负责封装数据集,StructureLoader负责将数据集按照序列长度聚类并批量加载数据。 - 文件位置:
ProteinMPNN/training/utils.py和ProteinMPNN/protein_mpnn_utils.py
2. make_fixed_positions_dict.py
- 简介:根据输入的 PDB 解析文件和指定的链及位置信息,生成固定位置的字典。
- 文件位置:
ProteinMPNN/helper_scripts/make_fixed_positions_dict.py
3. parse_multiple_chains.py
- 简介:解析包含多个蛋白质链的 PDB 文件,提取序列和坐标信息,并保存为 JSON 文件。
- 文件位置:
ProteinMPNN/helper_scripts/parse_multiple_chains.py
4. assign_fixed_chains.py
- 简介:根据输入的 JSON 文件和指定的链列表,分配固定链和设计链。
- 文件位置:
ProteinMPNN/helper_scripts/assign_fixed_chains.py
5. make_bias_AA.py
- 简介:根据输入的氨基酸列表和偏置列表,生成氨基酸偏置字典。
- 文件位置:
ProteinMPNN/helper_scripts/make_bias_AA.py
6. TMalign
- 简介:计算两个蛋白质链之间的 TM 分数、RMSD 等指标。
- 文件位置:
ProteinMPNN/training/parse_cif_noX.py
7. training.py
- 简介:用于训练蛋白质序列预测的神经网络模型。
- 文件位置:
ProteinMPNN/training/training.py
脚本使用
作者还提供了很多示例脚本,展示了如何使用 ProteinMPNN 进行不同场景下的蛋白质序列设计。
1. submit_example_1.sh
- 功能:简单单体示例,用于处理单个蛋白质链的设计。
- 步骤:
- 解析
inputs/PDB_monomers/pdbs/目录下的 PDB 文件为jsonl格式。 - 运行
protein_mpnn_run.py脚本进行序列设计,每个目标生成 2 条序列,采样温度为 0.1,随机种子为 37,批次大小为 1。
- 解析
- 结果
- 成功为两个蛋白质结构生成了序列,并记录了生成时间和序列长度。
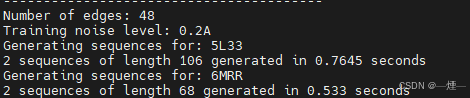
- 其中,5L33的结果

- 生物学意义
submit_example_1.sh 可以为单体蛋白质生成新的氨基酸序列,这些序列可能会改变蛋白质的催化活性、结合亲和力、稳定性等性质。例如,对于一种酶蛋白,通过设计新的序列可能提高其催化效率,从而更高效地参与生物化学反应。
2. submit_example_2.sh
- 功能:简单多链示例,处理包含多个蛋白质链的复合物。
- 步骤:
- 解析
inputs/PDB_complexes/pdbs/目录下的 PDB 文件为jsonl格式。 - 指定设计链为
A和B,并将设计信息保存到assigned_pdbs.jsonl中。 - 运行
protein_mpnn_run.py脚本进行序列设计,参数与示例 1 类似。
- 解析
- 结果
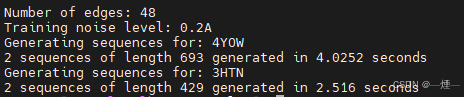
3. submit_example_3.sh
- 功能:直接从单个 PDB 文件路径进行设计。
4. submit_example_3_score_only.sh
- 功能:仅返回模型的得分(模型的不确定性),不进行序列设计。
5. submit_example_3_score_only_from_fasta.sh
- 功能:从 FASTA 文件加载序列,仅返回模型的得分(模型的不确定性)。
6. submit_example_4.sh
- 功能:固定某些残基位置,不进行设计。
7. submit_example_4_non_fixed.sh
- 功能:指定需要设计的残基位置。
8. submit_example_5.sh
- 功能:将某些位置绑定在一起(考虑对称性),即这些位置的氨基酸选择是相关的。
9. submit_example_6.sh
- 功能:同源寡聚体示例,处理由相同亚基组成的蛋白质复合物。
10. submit_example_7.sh
- 功能:返回序列的无条件概率(类似 PSSM)。
11. submit_example_8.sh
- 功能:添加氨基酸偏好,使某些氨基酸更有可能被选择。
12. submit_example_pssm.sh
- 功能:在设计序列时使用 PSSM(位置特异性得分矩阵)进行偏置。
这些示例脚本涵盖了 ProteinMPNN 的常见使用场景,用户可以根据自己的需求参考这些示例进行蛋白质序列设计。
测试
使用RFdiffusion生成的结构(序列全是G,150 AA)
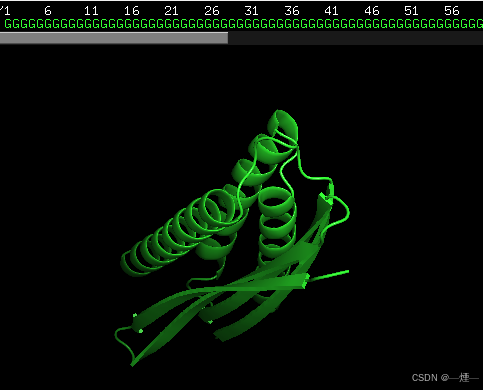
修改并运行submit_example_1.sh,结果:

- 温度(T):
T=0.1表示在生成序列时使用的采样温度为 0.1。较低的温度会使模型更倾向于选择概率较高的氨基酸,生成的序列相对保守;较高的温度会增加随机性,生成更多样化的序列。 - 样本编号(sample):
sample=1和sample=2分别表示生成的第 1 个和第 2 个样本序列。 - 得分(score):根据文档,
score是对设计的氨基酸进行负对数概率平均后的结果。负对数概率是衡量模型对生成序列的置信度,得分越低表示模型对生成的氨基酸序列越有信心。 - 全局得分(global_score):
global_score是对所有链中所有残基(包括固定和设计的残基)的负对数概率平均。同样,得分越低表示模型对整个结构 - 序列的匹配越有信心。 - 序列恢复率(seq_recovery):
seq_recovery表示生成的序列与原始序列的相似程度。在这个例子中,两个样本的序列恢复率均为 0.0333,说明生成的序列与原始序列的相似度非常低。


















 1265
1265

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









