






生物信息学数据分析流程的搭建是一项繁重而复杂的工作。随着行业的发展,各种生信流程框架层出不穷,比如有:
Nextflow
Snakemake
CWL
WDL
各种标准,各种规则,令人眼花缭乱。选择太多,往往令人无所适从。特别是新进入行业的人,不知道从何着手,一个一个学过去,对时间精力的耗费是巨大的,也是不必要的。不仅如此,既有的流程框架为了追求大而全,往往显得很笨重,这无疑增加了开发、维护和使用的难度。
针对现有流程框架的不足,我们开发了zflow,旨在为组学数据分析提供一个通用而又简单易用的轻量级框架。
https://github.com/jianzuoyi/zflow功能简介
支持单样本流程搭建:如转录组标准分析、WGS、WES重测序等。
支持配对样本流程搭建:如肿瘤NGS变异检测等。
支持加测:即支持一个样本多个文库,一个文库多条Lane的实验设计。
设计理念
我们相信 Linux 是生信流程设计的通用语言,坚持直接用Linux shell脚本编写生信流程。
整体框架
流程主要由两部分组成:
模板文件:我们将流程所需的所有软件/脚本调用,计算资源(Cpu, 内存)以及任务之间的依赖关系,在一个 XML 模板文件中定义。
解析器:zflow,负责将XML模板文件解析为Shell脚本。
投递器:针对不同的集群环境,编写相应的任务投递插件。
zflow程序只有一个,即所有流程通用的。而每一条流程,会编写一个XML模板文件。任务投递,则是根据集群类型的不同,交给不同的投递插件。
软件依赖
Python 3.9.18
pandas 2.0.3
networkx 3.2.1
安装好Python和依赖的软件包,并将Python所在的路径加入环境变量中,如:
export PATH=/path/to/python3.9.18/bin:$PATH快速开始
git clone git@github.com:jianzuoyi/zflow.git
cd zflow
./zflow -husage: zflow [-h] --xml XML [--env ENV] [--bed BED] [--par PAR] --config CONFIG --outdir OUTDIR [--overwrite] [--verbose]
A pipeline framework for NGS data analysis.
optional arguments:
-h, --help show this help message and exit
--xml XML XML config file for different project Type
--env ENV The path of Environment.sh [$PIPEDIR/config/Environment.sh]
--bed BED The path of BED file [Default is $Target in Environment.sh]
--par PAR Parameters file [Para=Value per line]
--config CONFIG Config file of Data path and Custom Infomation
--outdir OUTDIR Output directory
--overwrite Overwrite output results
--verbose Show detail message使用说明
1. 生成任务
准备config文件:config.tsv
Project Patient Sample Type Data
mRNA hg002_gm24385 hg002_gm24385 . /ifs/public/test-data/giab/hg002_gm24385.mrna.R[12].fastq.gz
mRNA hg002_gm26105 hg002_gm26105 . /ifs/public/test-data/giab/hg002_gm26105.mrna.R[12].fastq.gz
mRNA hg002_gm27730 hg002_gm27730 . /ifs/public/test-data/giab/hg002_gm27730.mrna.R[12].fastq.gz
mRNA hg004_gm24143 hg004_gm24143 . /ifs/public/test-data/giab/hg004_gm24143.mrna.R[12].fastq.gz
mRNA hg005_gm24631 hg005_gm24631 . /ifs/public/test-data/giab/hg005_gm24631.mrna.R[12].fastq.gzProject: 项目名称,可以任意设置
Patient: 病人/对象ID,可以重复,因为一个病人/对象可能有多个样本
Sample: 样本ID,一个病人/对象可以有多个样本,但样本ID必须唯一
Type:样本类型,用于区分配对样本,如果T代表肿瘤样本,N代表正常对照样本
Data: 数据路径。支持双端测序数据的分析。
准备运行脚本:run.sh
#!/bin/bash
set -euo pipefail
config=./config.tsv
outdir=./result
template=./RNAseq_Standard_Analysis_PE.xml
./zflow \
--xml $template \
--config $config \
--outdir $outdir \
--verbose--xml 流程模板文件
--config 样本信息配置文件
--outdir 分析结果输出目录
--verbose 解析XML过程中输出详细提示信息,否则采用安静模式
运行:
./run.sh最后输出:
...
Reading tasks
Checking task dependencies
Sorting tasks
Sorting tasks done
Local tasks: /nas/develop/zflow/result/Tasks/mRNA/bin/mRNA_tasks.sh最终输出的文件 mRNA_tasks.sh 即是总的Shell脚本文件,该文件中每一行是一个任务脚本。可以看到,每个任务脚本的文件名是以01,02这样的数字编号开头的,这些编号即表示该任务在整个流程中的顺序。
less mRNA_tasks.sh/nas/develop/zflow/result/Tasks/mRNA/bin/01_Begin_mRNA.sh
/nas/develop/zflow/result/Tasks/mRNA/bin/02_Cat_Fastq_hg002_gm24385_hg002_gm24385.sh
/nas/develop/zflow/result/Tasks/mRNA/bin/02_Cat_Fastq_hg002_gm26105_hg002_gm26105.sh
/nas/develop/zflow/result/Tasks/mRNA/bin/02_Cat_Fastq_hg002_gm27730_hg002_gm27730.sh
/nas/develop/zflow/result/Tasks/mRNA/bin/02_Cat_Fastq_hg004_gm24143_hg004_gm24143.sh
/nas/develop/zflow/result/Tasks/mRNA/bin/02_Cat_Fastq_hg005_gm24631_hg005_gm24631.sh
/nas/develop/zflow/result/Tasks/mRNA/bin/03_Cat_Fastq_hg002_gm24385.sh
/nas/develop/zflow/result/Tasks/mRNA/bin/03_Cat_Fastq_hg002_gm26105.sh
/nas/develop/zflow/result/Tasks/mRNA/bin/03_Cat_Fastq_hg002_gm27730.sh
/nas/develop/zflow/result/Tasks/mRNA/bin/03_Cat_Fastq_hg004_gm24143.sh
/nas/develop/zflow/result/Tasks/mRNA/bin/03_Cat_Fastq_hg005_gm24631.sh
/nas/develop/zflow/result/Tasks/mRNA/bin/04_FastQC_hg002_gm24385_hg002_gm24385.sh
/nas/develop/zflow/result/Tasks/mRNA/bin/04_FastQC_hg002_gm26105_hg002_gm26105.sh
/nas/develop/zflow/result/Tasks/mRNA/bin/04_FastQC_hg002_gm27730_hg002_gm27730.sh
/nas/develop/zflow/result/Tasks/mRNA/bin/04_FastQC_hg004_gm24143_hg004_gm24143.sh
/nas/develop/zflow/result/Tasks/mRNA/bin/04_FastQC_hg005_gm24631_hg005_gm24631.sh
/nas/develop/zflow/result/Tasks/mRNA/bin/04_Fastp_hg002_gm24385.sh
/nas/develop/zflow/result/Tasks/mRNA/bin/04_Fastp_hg002_gm26105.sh
/nas/develop/zflow/result/Tasks/mRNA/bin/04_Fastp_hg002_gm27730.sh
/nas/develop/zflow/result/Tasks/mRNA/bin/04_Fastp_hg004_gm24143.sh
/nas/develop/zflow/result/Tasks/mRNA/bin/04_Fastp_hg005_gm24631.sh
...2. 投递任务
理论上来说,逐行执行 mRNA_tasks.sh 中的脚本,就可以得到分析结果,只是不能充分利用集群的计算资源。
你可以将这些任务手动投递到集群上,编号相同的任务可以同时投递。这是一种解决方案,但显然不是最优的。更好的方案是写一个插件与zflow衔接,将zflow生成的任务自动投递到集群上。
这得益于zflow两点独有的设计:其在生成脚本时,同时保留了任务的依赖关系,以及每一个任务所需要的Cpu和内存资源。
mRNA_tasks.sh文件的后面,还跟着一些以#order开头的行,这些行就表示任务之间的依赖关系。后续插件可以根据这些信息将所有任务添加到一个DAG图当中,从而自动高效地向集群投递任务。
#order 01_Begin_mRNA before 02_Cat_Fastq_hg002_gm24385_hg002_gm24385
#order 01_Begin_mRNA before 02_Cat_Fastq_hg002_gm26105_hg002_gm26105
#order 01_Begin_mRNA before 02_Cat_Fastq_hg002_gm27730_hg002_gm27730
#order 01_Begin_mRNA before 02_Cat_Fastq_hg004_gm24143_hg004_gm24143
#order 01_Begin_mRNA before 02_Cat_Fastq_hg005_gm24631_hg005_gm24631
#order 02_Cat_Fastq_hg002_gm24385_hg002_gm24385 before 03_Cat_Fastq_hg002_gm24385
#order 02_Cat_Fastq_hg002_gm26105_hg002_gm26105 before 03_Cat_Fastq_hg002_gm26105
#order 02_Cat_Fastq_hg002_gm27730_hg002_gm27730 before 03_Cat_Fastq_hg002_gm27730
#order 02_Cat_Fastq_hg004_gm24143_hg004_gm24143 before 03_Cat_Fastq_hg004_gm24143
#order 02_Cat_Fastq_hg005_gm24631_hg005_gm24631 before 03_Cat_Fastq_hg005_gm24631
#order 03_Cat_Fastq_hg002_gm24385 before 04_FastQC_hg002_gm24385_hg002_gm24385
#order 03_Cat_Fastq_hg002_gm26105 before 04_FastQC_hg002_gm26105_hg002_gm26105
#order 03_Cat_Fastq_hg002_gm27730 before 04_FastQC_hg002_gm27730_hg002_gm27730
#order 03_Cat_Fastq_hg004_gm24143 before 04_FastQC_hg004_gm24143_hg004_gm24143
#order 03_Cat_Fastq_hg005_gm24631 before 04_FastQC_hg005_gm24631_hg005_gm24631
#order 03_Cat_Fastq_hg002_gm24385 before 04_Fastp_hg002_gm24385
#order 03_Cat_Fastq_hg002_gm26105 before 04_Fastp_hg002_gm26105
#order 03_Cat_Fastq_hg002_gm27730 before 04_Fastp_hg002_gm27730
#order 03_Cat_Fastq_hg004_gm24143 before 04_Fastp_hg004_gm24143
#order 03_Cat_Fastq_hg005_gm24631 before 04_Fastp_hg005_gm24631
...你可能要问,那每一步的计算资源(Cpu,内存)需求在哪里呢?可以打开任意一个脚本,如:
less /nas/develop/zflow/result/Tasks/mRNA/bin/05_Align_Hisat2_hg002_gm24385.sh#!/bin/bash
set -euxo pipefail
source /pub/pipeline/RNAseq/config/Environment.sh
Cpu=8
Mem=32
cd /nas/develop/zflow/result/mRNA/hg002_gm24385/hg002_gm24385/02.Aln
hisat2 \
-x $REFERENCE_GENOME \
-1 ../01.QC/hg002_gm24385_clean_R1.fq.gz \
-2 ../01.QC/hg002_gm24385_clean_R2.fq.gz \
--summary-file hg002_gm24385_summary.txt \
--new-summary \
-p 8 \
--dta -t |\
samtools view -Sb > hg002_gm24385.bam
ln -s `pwd`/hg002_gm24385_summary.txt /nas/develop/zflow/result/mRNA/multiqc/multiqc_WDir/hisat2/可以看到,脚本的前面有:Cpu=8, Mem=32,即是指示该步骤需要8条线程,32G内存。负责投递任务的插件可以读取这两个信息用于任务投递。顺便提一下,任务脚本开始有一行:
source /pub/pipeline/RNAseq/config/Environment.sh即是导入环境变量,任务中所需的软件的路径应该在这个Environment.sh文件中定义好,如本任务所需要的 hisat2, samtools应该在Environment.sh文件中将其所在的目录加入PATH。
最后来看一下脚本目录的全家福:
01_Begin_mRNA.sh
02_Cat_Fastq_hg002_gm24385_hg002_gm24385.sh
02_Cat_Fastq_hg002_gm26105_hg002_gm26105.sh
02_Cat_Fastq_hg002_gm27730_hg002_gm27730.sh
02_Cat_Fastq_hg004_gm24143_hg004_gm24143.sh
02_Cat_Fastq_hg005_gm24631_hg005_gm24631.sh
03_Cat_Fastq_hg002_gm24385.sh
03_Cat_Fastq_hg002_gm26105.sh
03_Cat_Fastq_hg002_gm27730.sh
03_Cat_Fastq_hg004_gm24143.sh
03_Cat_Fastq_hg005_gm24631.sh
04_FastQC_hg002_gm24385_hg002_gm24385.sh
04_FastQC_hg002_gm26105_hg002_gm26105.sh
04_FastQC_hg002_gm27730_hg002_gm27730.sh
04_FastQC_hg004_gm24143_hg004_gm24143.sh
04_FastQC_hg005_gm24631_hg005_gm24631.sh
04_Fastp_hg002_gm24385.sh
04_Fastp_hg002_gm26105.sh
04_Fastp_hg002_gm27730.sh
04_Fastp_hg004_gm24143.sh
04_Fastp_hg005_gm24631.sh
05_Align_Hisat2_hg002_gm24385.sh
05_Align_Hisat2_hg002_gm26105.sh
05_Align_Hisat2_hg002_gm27730.sh
05_Align_Hisat2_hg004_gm24143.sh
05_Align_Hisat2_hg005_gm24631.sh
05_FastQC2_hg002_gm24385.sh
05_FastQC2_hg002_gm26105.sh
05_FastQC2_hg002_gm27730.sh
05_FastQC2_hg004_gm24143.sh
05_FastQC2_hg005_gm24631.sh
06_Align_Hisat2_Sort_hg002_gm24385.sh
06_Align_Hisat2_Sort_hg002_gm26105.sh
06_Align_Hisat2_Sort_hg002_gm27730.sh
06_Align_Hisat2_Sort_hg004_gm24143.sh
06_Align_Hisat2_Sort_hg005_gm24631.sh
07_Stringtie_hg002_gm24385.sh
07_Stringtie_hg002_gm26105.sh
07_Stringtie_hg002_gm27730.sh
07_Stringtie_hg004_gm24143.sh
07_Stringtie_hg005_gm24631.sh
08_Stringtie_preDE_mRNA.sh
09_MultiQC_mRNA.sh
10_End_mRNA.sh
mRNA_tasks.sh一个总的脚本文件mRNA_tasks.sh,记录所有要执行的任务以及任务之间的依赖关系。
多个任务脚本文件,记录着每一个任务要执行的命令以及所需要的计算资源。
模板简介
流程的描述信息我们统一放到一个XML模板文件当中。该模板的结构为:
<Pipeline>
<Task>
<Name>Align_Hisat2_$Sample</Name>
<Rely>Fastp_$Sample</Rely>
<Cpu>8</Cpu>
<Mem>32</Mem>
<Workdir>$Outdir/$Project/$Patient/$Sample/02.Aln</Workdir>
<Command><![CDATA[
hisat2 \
-x $REFERENCE_GENOME \
-1 ../01.QC/$Sample_clean_R1.fq.gz \
-2 ../01.QC/$Sample_clean_R2.fq.gz \
--summary-file $Sample_summary.txt \
--new-summary \
-p $Cpu \
--dta -t |\
samtools view -Sb > $Sample.bam
ln -s `pwd`/$Sample_summary.txt $Outdir/$Project/multiqc/multiqc_WDir/hisat2/
]]></Command>
</Task>
<ENV>
<Environment>/path/to/RNAseq/config/Environment.sh</Environment>
</ENV>
</Pipeline>每一个配对的<Task>,</Task>标签内是一个任务的描述。其中包含:
Name:任务名称
Rely:任务依赖的步骤名称
Cpu:所需Cpu资源
Mem:所需内存资源
Workdir:工作目录
Command: 任务命令
其中以美元符号$或者@开头的为变量($表示普通变量,@表示数组),在任务解析过程中将会被zflow依据输入的参数信息替换为具体的值。详情请参考流程示例文件:RNAseq_Standard_Analysis_PE.xml。
Environment.sh文件的作用前述已提及,用于放置所有环境变量。
流程设计
流程搭建非常容易,直接在XML中写Shell代码就可以了。但是zflow流程有一些保留的变量,或者说关键字,需要用户事先掌握。请放心,关键字的数量并不多,并且都非常容易理解。
XML标签中的关键字:
Name, Rely, Cpu, Mem, Workdir, Command,各关键字的作用前述也提及。
来源于命令行的关键字:
$Config:配置文件,即zflow的--config参数所指定的文件
$Target:BED格式文件,如WES或Panel测序中的探针捕获区域文件
$Outdir:输出目录
来源于配置文件中的关键字:
总的来说,配置文件中的每一列,会成为一个变量传递给XML,另外一些数组可以通过计算变换获得。
$Project:项目名称
$Patient:病人/对象ID
:样本,如果是肿瘤样本,可以是Tumor表示肿瘤样本ID, $Normal表示正常样本ID
$Datadir:测序数据的原始位置
$Lib:文库名称
$Lane:Lane名称
@Lib:数组,代表某一样本的所有文库
@Lane:数组,代表某一文库的所有Lane
@Sample:数据,代表所有样本ID
@Tumor:数组,代表所有肿瘤样本ID
@Normal:数组,代表所有正常样本ID
需要说明的是,配置文件中列出的5个列是强制要求的:Project Patient Sample Type Data,除此之外,你还可以在其中增加列,这些列名同样会作为变量传递给XML。
更多变量想传递进XML,还可以通过 zflow 的 --par 参数实现:
--par PAR Parameters file [Para=Value per line]在这个参数文件中,每一行定义一个变量,其格式为:
参数名称=参数对应的值(可以是某个数值,或者文件的路径等)可以通过配置文件config.tsv和专门的参数文件--par灵活地向XML文件传递变量,这有效地保证了zflow的灵活性。比如,你可以在config.tsv文件中增加一列,表示样本分组。也可以将分组信息单独放在一个文件中,再在--par文件中指明分组文件所有的路径,这都是非常灵活的。
特点总结
综上可以看出zflow设计生信流程的特点:
简单易用。流程的所有设计都在XML文件中,zflow只负责解析XML,针对不同的流程,zflow无须做任何更改。
轻量级。zflow由单个Python脚本构成,仅有600多行代码。
轻松解决任务之间的依赖关系。zflow能够在生成任务脚本的过程中顺便生成任务之间的依赖关系。开发人员所需要做的工作仅仅是在XML文件中指定一下某个任务需要依赖于哪个或哪些任务即可。更进一步,如果任务关系指定错误,zflow能够给出错误提醒。
模块化设计。zflow被设计成只负责生成Shell脚本,后续的集群任务投递交给专用的插件完成。这极大地保证了流程框架的可扩展性。
很多生信流程,值得用zflow重写一遍。
许可证
zflow 支持学术免费使用,欢迎 Fork、加星和下载。如需商用,请联系【简说基因】获得授权。
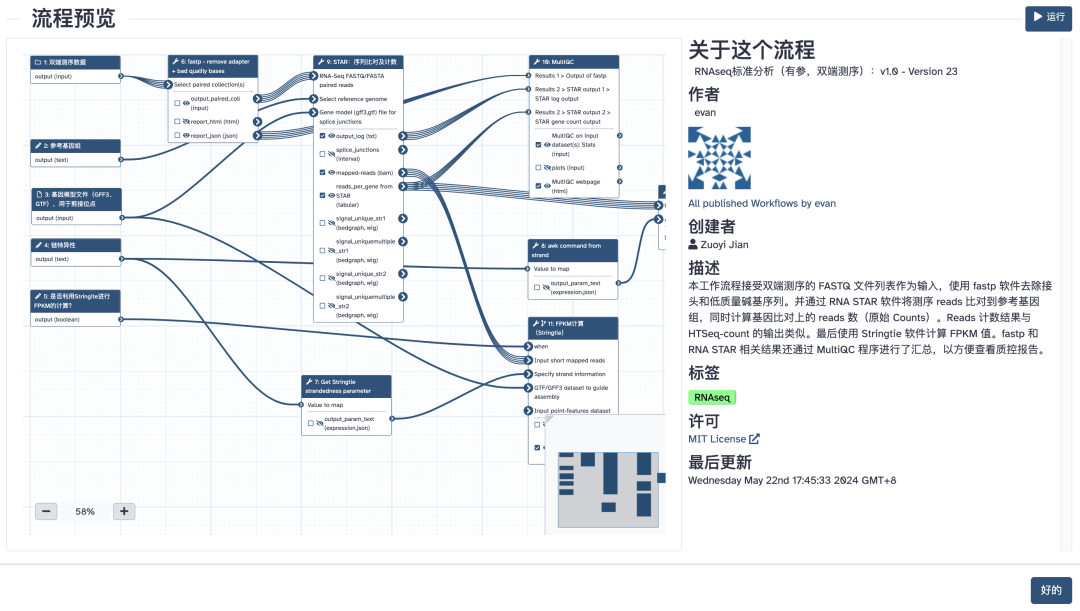
云上转录组分析流程(点击图片跳转)
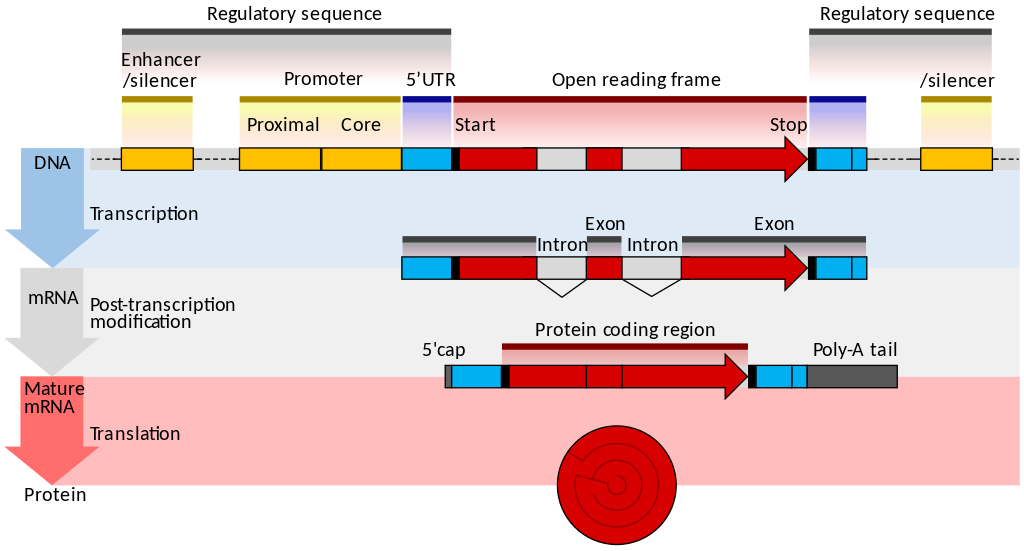
一文读懂scRNA-seq数据分析(点击图片跳转)
如何自学生物信息学:从菜鸟到专家(点击图片跳转)

往期精彩:
网上最全的 R 语言图库(建议收藏)| 简说基因 Recommend
生物信息学必备的R语言相关参考书 | 简说基因 Recommand
关于简说基因
生信平台
Galaxy中国(UseGalaxy.cn)致力于打造中国人的云上生物信息基础设施。大量在线工具免费使用。无需安装,用完即走。活跃的用户社区,随时交流使用心得。
联系方式
QQ交流群(免费):925694514
微信交流群(免费):加微信好友,注明“Galaxy交流群”
客服微信:usegalaxy















 1677
1677

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









