






从今天开始,用 21 天精通单细胞数据分析。我们将理论联系实际,边学边练,本文先从理论开始。
https://usegalaxy.cn/training-material/topics/single-cell/tutorials/scrna-intro/slides_ZH-CN.html#1
在线 PPT 有更好的阅读体验,请通过上述链接查看。
提示: 在线阅读,按 P 显示演讲者提示;方向箭头切换幻灯片。





• 大家好,欢迎来到银河系单细胞 RNA 测序分析研讨会。
• 在这里,我们将向您介绍处理单细胞数据时的一些基础知识和概念。

• 我们首先来探讨一下批量 RNA 测序和单细胞 RNA 测序数据之间的区别。
• 使用批量 RNA 测序,我们通过观察每个组织中检测到的每个基因的平均表达来比较两种组织。
• 由于考虑的 RNA 分子数量,测序深度和分析强度相当高。
• 差异表达随后被测量为一个给定基因在一种组织与另一种组织之间的相对表达。

• 通过单细胞 RNA 测序分析,研究的焦点从测量组织的平均表达转移到了个体细胞层面。
• 并朝着测量这些组织中单个细胞的特定基因表达。
• 在这里,我们不再比较组织与组织,而是细胞与细胞。
• 每个细胞被赋予一个基因表达谱,该表达谱描述了在该细胞内检测到的基因的相对丰度。
• 许多细胞共享相同的基因谱,其中基因谱理想地描述了一种细胞类型。
• 有时我们需要比较不同组织中的单细胞数据集,我们发现许多组织中的细胞共享相同的细胞类型。
• 例如,看看那些在两种组织中共享的紫色和绿色基因表达谱。

• 新技术意味着新的方法和技术,用以利用随之而来的新特性。
• 单细胞 RNA 测序数据需要不同的文库制备、测序、质量控制和分析方法。

例如,细胞是如何被捕获和测序的?

在批量 RNA 测序分析中,该过程包括取样、去除不需要的分子以及对其余内容进行测序。

• 对于单细胞分析,过程基本相同,只是每个样本是一个细胞。
• 因此必须与其他细胞分开进行测序。
• 一旦分离,就会给每个细胞添加独特的条形码,然后进行测序。

• 单细胞的分辨率在细胞层面,每个细胞都是独一无二的。
• 因此,生物学重复的概念与批量 RNA-seq 中的并不完全相同。

细胞分离可以通过不同的方式进行。

一种方法是手动移液,其中湿实验室的科学家使用一根长细管吸起单个细胞。

他们可以这样做数百次来分离数百个细胞,但这容易出错,并且经常一起分离出多个细胞。

另一种方法是流式细胞术,这减少了这一阶段的人为误差成分。

• 流式细胞术将细胞悬浮在浅液浴中,并沿着一个狭窄的通道流动,通道的宽度刚好够一个细胞通过。
• 细胞可以通过多种属性进行筛选,例如通过它们的光散射特性和荧光细胞标记。
• 细胞可以通过这种方式被标记和分离。

• 光学散射特性可以用来探测细胞的大小和一致性,其中小于激光波长的细胞会产生较低的强度和更不一致的散射模式。
• 光学散射主要有两种类型:前向散射和侧向散射。

• 前向散射与主激光对齐,用于测量细胞直径,非常适合通过它们的大小轮廓来区分不同的细胞。
• 例如,单核细胞通常比淋巴细胞大,这在示例图像的 X 轴上可以看到。

侧向散射是垂直于主激光的,它测量细胞的粒度,非常适合区分内部结构不太明确的细胞,如示例图像 Y 轴上的粒细胞。

• 细胞也可以通过流式细胞术(FACS)根据其细胞表面标志物进行筛选和表征。
• 通过将不同的表面标记物强度相互对比,可以根据这些荧光特性对细胞进行分离、门控和标记。

• 一旦分离,细胞就可以进行条形码标记。
• 条形码是添加到每个 RNA 分子上的独特序列。
• 它们不是分子特有的,而是细胞特有的,以至于如果任何两个 RNA 分子存在于同一个细胞中,它们将被相同的细胞条形码标记。
• 来自不同细胞的 RNA 分子将具有不同的细胞条形码。

一旦 RNA 分子被细胞条形码标记,它们就可以被扩增,无论是单独还是一起混合,扩增后的产物与其原始对应物共享相同的细胞条形码。

• PCR 扩增基因产物,使其在测序过程中更易于检测。
• 当需要大量扩增基因产物,如常规 RNA 测序的情况,PCR 在以相当均衡的方式扩增所有产物方面表现良好。
• 然而,在单细胞产品的情况下,需要扩增的量非常小,在这个阶段可能会错过许多独特的读取,而其他读取可能会过度扩增,正如示例中的蓝色和红色转录本所示。

• 为了防止这种类型的扩增偏差,我们可以在条形码中加入一个随机元素。
• 这些被称为 UMIs 的随机条形码,唯一地标记转录本,使得同一基因的任意两个转录本很可能会有不同的随机条形码。

• 让我们考虑左侧的例子:细胞内有 2 个红色转录本和 2 个蓝色转录本,在扩增后分别等同于 6 个红色转录本和 3 个蓝色转录本。
• 如果我们仅通过观察放大的读取来比较红色和蓝色转录本之间的差异基因表达,我们会得出错误的结论,即红色转录本的表达量是蓝色的两倍。

然而,如果我们按它们的 UMIs 对读取进行分组,然后只计算每个转录本的唯一 UMIs 的数量,对共享相同转录本和 UMI 的读取进行去重,我们得到 2 个红色读取和 2 个蓝色读取,这更好地代表了转录本的真实数量。

• UMIs 相对随机,但并非真正的随机。
• 请注意,粉红色的 UMI 出现了两次:一次在蓝色转录本中,一次在红色转录本中。

这是由于转录本的数量通常比可用的 UMI(唯一分子标识符)多,这两者都取决于细胞中的转录本数量和条形码的长度。

• 考虑一组长度为 5 的条形码,相邻条形码之间的编辑距离为 1,以及另一组编辑距离为 2 的条形码。
• 前者对常见的 1 个碱基对的测序错误不具有鲁棒性,但后者仅允许使用一半数量的条形码。
• 这种在可用条形码数量与防范测序错误之间的权衡,在设计细胞条形码和唯一分子标识符(UMIs)时至关重要。

在扩增的背景下,唯一分子标识符(UMIs)不需要是唯一的,它们只需要足够随机,以便去重转录本,从而更准确地估计细胞内的转录本数量。

让我们简单回顾一下我们学到的内容:首先,每个细胞中的每个 RNA 分子都添加了细胞条形码。

• 然后我们为所有转录本添加随机的 UMIs(唯一分子标识符),这进一步标记了分子。
• 这些随后可以用来在扩增后去除转录本的重复。
• 扩增后我们需要进行一些质量控制。

• 实现这一目标的一种方法是为基因和细胞的检测限度设定阈值。
• 考虑一个仅由 3 个基因(G1、G2 和 G3)和 5 个细胞(A、B、C、D 和 E)控制的分析。
• 上表第一行定义了库的大小,即每个细胞中所有基因的信使 RNA 总数。
• 后续的行是基因可检测性的阈值,显示了在阈值从 0 到 4 的范围内,每个细胞中检测到的基因数量。
• 我们发现,即使在给定细胞中检测到的转录本阈值大于 3,分析中仍然保留了 3 个细胞:B、C 和 E。在下面的表格中,情况正好相反,展示了每个基因在所有细胞中的总转录本数量。
• 通过设定可检测性的阈值,我们可以看到有多少细胞被该阈值下的基因所描述。
• 在这两种情况下,我们都可以看到,如果我们将阈值设置得太低,那么我们就有保留低质量基因或细胞的风险;但如果我们将可检测性的阈值设置得太高,那么我们就有丢失太多细胞的风险。

• 然而,过滤可能是一种奢侈,因为许多单细胞 RNA 测序数据集通常与批量 RNA 测序相比具有较低的测序深度。
• 在标准化过程中,样本相互之间进行缩放,以使它们更具可比性。
• 这通常通过使用中位数值来完成。例如,在 DE-Seq 标准化中,取一个细胞的几何平均计数,然后该细胞中的每个基因值都除以它以及所有细胞的几何平均数的中位数值。
• 如果中位数基因表达水平较高,那么这种标准化方法的效果就非常好。

• 但如果中位数基因表达为零,正如单细胞数据中常见的情况,那么我们就会遇到除以零的问题。
• 有一些方法可以解决这些零计数的问题。

• 一种这样的方法是 Scran 方法,它通过创建重叠的细胞池来工作,使得任何单个细胞都通过具有相似文库大小的细胞来表征。
• 该方法涉及将所有细胞根据其文库大小分为奇数组和偶数组,并将它们排列在一个环形结构上,其中环上相邻的细胞具有相似的大小。

• 定义了固定大小的重叠池,结果是每个细胞被多个池定义。
• 然后可以通过它所在的池构建该细胞的线性模型,所有细胞的归一化因子也可以通过这种方式确定。

• 通过这种方法,通过将具有低库大小的细胞转化为可应用于类似细胞的大小因子的有用组成部分,巧妙地解决了低序列覆盖率的问题。
• 这种新颖的标准化方法在几年前很常见,但随着测序技术的进步,矩阵中许多零计数的问题变得不那么重要了,我们又可以再次使用批量 RNA-seq 方法来推导标准化大小因子。

• 在进行单细胞 RNA 分析时,我们需要考虑的其他因素是可能混淆分析的非目标因素。
• 理想情况下,我们希望看到区分不同类型细胞的基因表达谱是由生物学变异驱动的。
• 然而,来自技术和生物学来源的混杂变异对分析没有用处,但却对变异性有所贡献。

• 混杂的生物学变异以两种形式出现:转录爆发和细胞周期变化。
• 转录爆发是一种在细胞中发生的现象,其中转录发生在活跃和不活跃的离散状态之间,这些状态之间的间隔很难建模。
• 在批量 RNA 测序中,这种现象是不易察觉的,因为影响会在许多细胞上平均化。但在单细胞中,即使是同一类型的两个细胞也可能表现出不同的基因谱,仅仅因为一个细胞正在积极转录,而另一个则没有。
• 这不是我们可以在分析中控制的东西,但在理解为什么细胞簇可能存在噪声时,我们应该意识到这一点。
• 另一方面,细胞周期的变化是一个更为人们所熟知的过程,其中一种类型细胞中的 RNA 含量大约是同类型细胞的两倍,这是因为一个细胞处于细胞周期的早期 G1 阶段,而另一个细胞处于 M 期。
• 有些基因已知会与细胞周期共变,因此通过回归这些基因的影响,我们可以控制细胞周期的影响。

• 技术变异以三种形式出现:扩增偏差、丢失事件和文库大小变异。
• 扩增偏差可以通过 UMIs(唯一分子标识符)来减轻,这一点之前已经展示过。
• 零值事件导致计数矩阵中普遍存在的零,通过使用巧妙的标准化技术,例如之前展示的池化方法,以及使用更好的测序方法,可以减少它们的影响。

• 文库大小的变化源于多种不同的原因,但它是分析中变异的主要来源。
• 与批量 RNA 测序一样,通过良好的标准化方法可以减少这种偏差。

• 一旦我们从分析中移除了不想要的混杂因素,我们就面临量化细胞间关系的问题。
• 从数据分析的角度来看,我们将每个细胞视为一个观测值,每个基因视为一个变量。
• 对于大型基因组来说,这意味着数据集的维度非常高。细胞在这个高维空间中稀疏分布,成为其中的点,这使得识别自然的群体分类变得困难。
• 通过简单地过滤掉在所有细胞中似乎不表现出差异表达的基因,高维空间可以大大减少。
• 要找出这些细胞之间的关系,我们需要定义细胞之间的距离。

• 距离矩阵正是如此,它通过一个单一的得分来定义任意两个细胞之间的距离。
• 在这里,我们使用三维数据集(包含 3 个基因 G1、G2 和 G3,以及 3 个细胞 R、P 和 V)上的欧几里得距离。
• 任意两个细胞之间的距离可以计算为基因值差异的平方和。
• 注意距离矩阵沿对角线是对称的,这证实了例如从 R 细胞到 V 细胞的距离与从 V 细胞到 R 细胞的距离相同,正如预期的那样。

• 一旦生成了距离矩阵,我们可以在距离矩阵上执行 K 最邻近算法,其中在细胞之间生成有向边。
• 对于距离矩阵的每一行,选择 K 个具有最小距离值的细胞,代表当前行的细胞与所选列细胞最近的邻居。
• 如果相邻细胞之间共享边,则这种方法称为共享最邻近方法。

• 我们可以将这个三维空间轻松地表示为 3 个独立的轴,轴上的点表示细胞。
• 将这个相对较低维度的示例集外推到一个具有数千维度的真实数据集,超出了人类能力的范围。
• 降维是一种技术,它将高维数据集转换成低维表示,通常是二维的,试图保留数据点之间的距离。
• 在这里,细胞之间的相对差异在高维和低维表示中都得到了保留。
• 存在许多不同种类的降维技术,每种技术都有其自身的优势和劣势,这取决于数据的类型和维度。

• 一旦数据集的变量数量通过过滤和降维被充分减少,聚类就可以更容易地进行。
• 在这个二维投影图中,每个圆圈代表一个细胞,独特的颜色表示它们被分配到的簇。
• 这些彩色细胞群之间的物理距离告诉我们这种投影的聚类效果有多好。

• 通过检查每个簇中相对于所有其他簇表达差异最大的基因,可以找到描述该簇所代表细胞类型的线索。
• 细胞类型通常由特定标记基因的表达来表征,这些基因的存在是类型的强烈指示。
• 标记基因的发现随后可以用来注释这些簇。

我们还可以进一步推导出这些簇之间的关系,通过计算基于每个簇中的噪声量来构建谱系树,期望干细胞具有嘈杂的表达谱,从而形成更宽的簇,而成熟细胞具有非常清晰的表达谱,从而形成更紧密的簇。

• 在分析中可能遇到的聚类类型取决于输入数据集,其中从晚期样本中提取的细胞不太可能聚集在一起,更有可能产生被称为硬聚类的大可见间隙,这些间隙清晰地定义了不同类型的细胞。
• 较早阶段的数据集更有可能产生较软的聚类,其中邻近的聚类共享柔软的边界,因为聚类之间略有交织。

软聚类是预期中的现象,因为尽管聚类是一种将数据离散分区的统计方法,但数据背后的细胞生物学是一个连续的过程,在这个过程中,细胞通过中间阶段从一个明确定义的状态过渡到另一个状态,这些中间阶段在两个簇中心之间得到表示。

• 由于这些单细胞数据集的连续性质以及数据的极高维度,离散分割通常是一个对数据进行分割的较差模型。
• 如果我们假设细胞簇通过过渡细胞相互关联,而这些过渡细胞自然位于簇之间,那么流形学习技术则更为适用。
• 这些技术衍生出一个表达景观,不仅可以用于比较不同簇之间的关系,还可以用于推断谱系和层级结构。
• 实际上进行聚类分析时,有三种常用的方法:K-均值法、层次聚类法和社区聚类法。

• K-均值和 K-中位数遵循相同的方法:簇的数量是预先定义的,并在随机位置初始化。
• 然后,根据更靠近这些位置而非其他位置的细胞的贡献来更新这些位置。
• 这个过程会多次进行,直到位置不再显著变化,或达到设定的迭代次数。
• 每个细胞的最终分配则成为聚类分配。

• 层次聚类更加灵活,不需要初始参数来定义结果聚类的数量。
• 在这里,距离矩阵中最近的两个点被合并为一个单一的组,重新计算距离,然后再次将最近的两个点合并。
• 这个过程会一直重复,直到所有数据都被整合为一个。
• 通过逆向追踪过程,可以建立一个由树状图表示的层次结构。

• Louvain 聚类是一种广泛用于单细胞数据的社区聚类方法。
• 在这里,每个细胞都被分配了一个自己的邻域,并且计算了邻域之间内部和外部链接的数量。
• 对于每一次迭代,随机选择一个细胞,并将其纳入另一个细胞的邻近区域,然后再次计算内部和外部链接。
• 如果新的配置减少了外部链接的数量,而增加了更多的内部链接,那么这个配置就会被保留。

如果新的配置反而增加了外部链接的数量,则该配置将被拒绝,并选择另一个单元格进行测试。通过多次执行此操作,可以构建出用户所需的特定程度的单元格社区结构。

• 单细胞分析并非易事,从过滤到标准化,再到降维和聚类,每个阶段都可能显著影响分析结果。
• 由于分析的可变性,面对不确定性时,人们不应感到恐慌。
• 目标是不断探索数据,直到它开始反映出生物学的实质。
• 这可能需要很多次尝试才能实现,而且可能永远无法达到完美,但想法是尽可能尝试不同的方法,看看你能得到哪些稳健的结论。




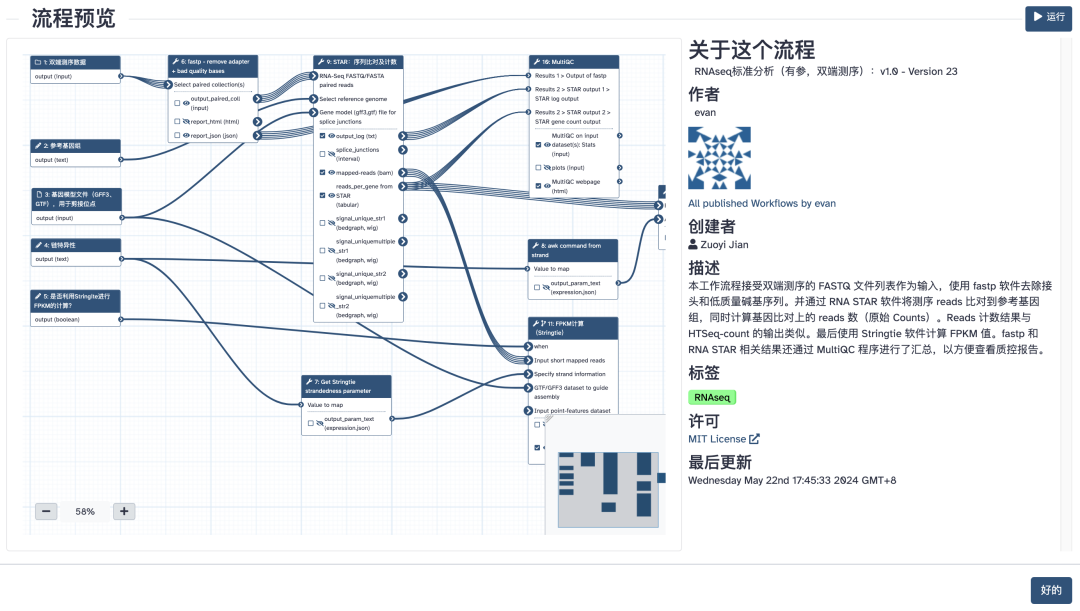
云上转录组分析流程(点击图片跳转)
什么生信流程语言让你极度爽?(点击图片跳转)

推荐阅读:
网上最全的 R 语言图库(建议收藏)| 简说基因 Recommend
生物信息学必备的R语言相关参考书 | 简说基因 Recommand
关于简说基因
生信平台
Galaxy中国(UseGalaxy.cn)致力于打造中国人的云上生物信息基础设施。大量在线工具免费使用。无需安装,用完即走。活跃的用户社区,随时交流使用心得。
联系方式
QQ交流群(免费):925694514
微信交流群(免费):加微信好友,注明“Galaxy交流群”
客服微信:usegalaxy















 1698
1698











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









