






ChIP-seq是一种结合位点分析法,用于研究体内蛋白质与DNA相互作用。通过染色质免疫共沉淀技术(ChIP)与第二代测序技术相结合,在全基因组范围内检测与组蛋白、转录因子等互作的DNA区域。
实验过程

流程简介

1. fastp:原始数据质控,将 Raw data 转换成 Clean data。
2. bowtie2:将经过质控的 Clean data 比对到参考基因组上,得到比对文件(BAM格式)。
3. picard:去除 BAM 文件中的 PCR 重复。
4. macs2:检出 ChIP-seq 峰。
5. MultiQC:汇总 fastp,bowtie2 以及 macs2 的统计结果。
运行流程
进入网站
https://usegalaxy.cn
找到流程

参数设置
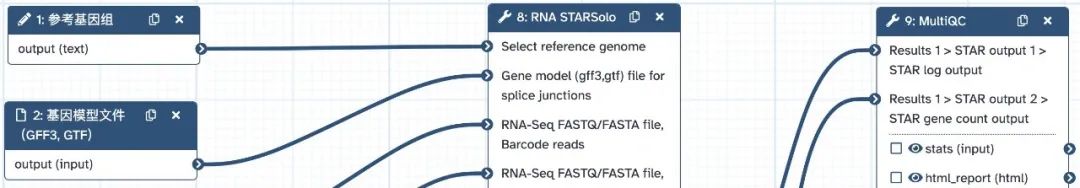
本流程支持 ChIP-seq 双端测序、有对照样本的数据分析,需要设置的参数如下图所示:

需要注意的是有效基因组大小,指的是基因组可比对(mappable)区域的大小。可以参照如下网站:
https://deeptools.readthedocs.io/en/develop/content/feature/effectiveGenomeSize.html
该网站上计算好了常见基因组的有效大小,也可以利用其介绍的工具自行计算。
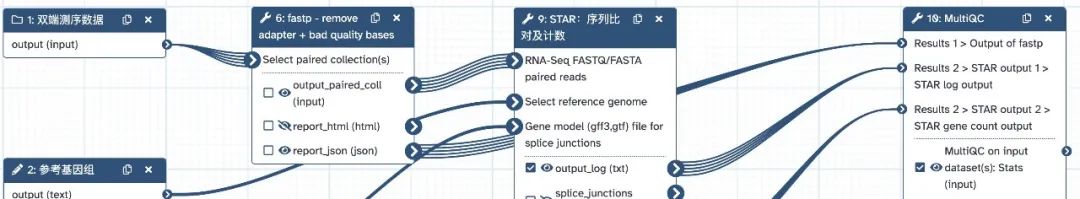
输出结果
fastp 输出:
• 质控结果 HTML (对照)
• 质控结果 HTML (实验)
bowtie2 输出:
• 比对统计(对照)
• 比对统计(实验)
picard 输出:
• 去重后的 BAM (对照)
• 去重后的 BAM (实验)
macs2 输出:
• 峰值文件(Narrow Peaks)
MultiQC 输出:
• 统计结果汇总 HTML(来源于 fastp,bowtie2,macs2)
注意事项
本流程采用 bowtie2 作为比对工具,目前 Galaxy 平台上构建了常见物种基因组的 bowtie2 索引文件,如果您需要的基因组索引不存在,欢迎联系我们添加。
一键分析10X单细胞数据(点击图片跳转)
一键分析Bulk转录组数据(点击图片跳转)
推荐阅读:
网上最全的 R 语言图库(建议收藏)| 简说基因 Recommend
生物信息学必备的R语言相关参考书 | 简说基因 Recommand
关于简说基因
生信平台
Galaxy中国(UseGalaxy.cn)致力于打造中国人的云上生物信息基础设施。大量在线工具免费使用。无需安装,用完即走。活跃的用户社区,随时交流使用心得。
联系方式
QQ交流群(免费):925694514
微信交流群(免费):加微信好友,注明“Galaxy交流群”
客服微信:usegalaxy


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









