







题目:SeqCode: a nomenclatural code for prokaryotes described from sequence data
期刊:Nature Microbiology
发表年月:2022年9月
第一作者:Brian P. Hedlund
通讯作者:Stephanus N. Venter & William B. Whitman
第一单位:美国内华达大学生命科学学院
影响因子:17.74
DOI:10.1038/s41564-022-01214-9
原文链接:https://www.nature.com/articles/s41564-022-01214-9
- 摘要 -
大多数原核生物不能通过纯培养获得,因此无法根据国际原核生物命名法 (ICNP) 的规则和建议进行命名。在本文中,我们总结了 SeqCode 的发展历程,这是一种基于基因组序列来命名的一种命名规则。该规则可基于已分离获得的基因组、宏基因组组装基因组或单扩增基因组序列有效地对原核生物进行命名。此外,它在名称的组成和优先级规则上与 ICNP 类似。SeqCode在 SeqCode Registry (https://seqco.de/) 上运行,通过该网站进行注册、查验名称和命名类型并链接到元数据。我们描述了 SeqCode 中当前可用于注册和验证名称(包括候选名称)的两种路径,并为两者提供了示例。我们还提供了关于 DNA 序列最低标准的建议。因此,SeqCode 不考虑其可培养性并为所有原核生物的命名提供了一个可重复且客观的框架,促进了微生物学科之间的交流。
- 引言 -
人们普遍认为,《国际原核生物命名法》(ICNP)要求将无污染和可传代培养的生物作为可命名的类型,这阻碍了对未培养和难培养的原核生物(古生菌和细菌)的命名,从而阻碍了微生物多样性的有效交流。迄今,尚未培养的类群约占原核生物系统发育多样性的85%,而已命名的原核生物占总物种的不到0.2%。当排除未培养的大多数微生物,生命树的很大一部分被降级为秩序不佳、模棱两可且通常是同义词的名称或字母数字。这些字母数字命名原则大都不利于记忆,因为每个字母或数字都导致了记忆或数字范围的限制。而分类学的名称应该通过一个单词来记住,特别是当其名称本身具有意义或为人们所熟知。
为解决这一问题,Konstantinidis等人和后来的Murray等人提出了两个方案(详见Roadmap for naming uncultivated Archaea and Bacteria | Nature Microbiology),得到了来自22个国家和六大洲的121位作者和署名人的赞同。最初的“Plan A”是基于DNA序列可作为命名类型并纳入现有的ICNP基础设施的提议。而国际原核生物系统学委员会(ICSP)拒绝了这一提议,因此“Plan B”得以落地,该计划呼吁制定新的命名规则。
- 方法 -
公共宣传和建立共识
该项目花费了大量精力与科研界进行沟通,以便微生物系统学的前进道路上建立共识。为取得共识,项目组举办了四大系列研讨会。
第一次研讨会
2018/10
第一个推广工作是由三部分组成的线上研讨会,题为 "Microbial systematics for the next decade",于2018年10月举行。该研讨会的目的是让不同的利益相关者参与讨论影响微生物系统学前景的关键问题。每个研讨会包括两个15分钟的演讲,然后是15分钟的四至五人分组讨论,以及15分钟的分组报告。研讨会后的调查问卷收集了对原核生物系统学未来发展方向的一般问题的回答。为了最大限度地提高“生产力”,所有参与者都有阅读任务,并被要求在每次研讨会前提出讨论的意见和想法。为了确保观点的广泛性,发言者包括微生物系统学的专家和来自植物和原生动物分类学的相关领域。研讨会三个部分的主题如下。(1)名称中应包含什么? 正式的分类学命名规则的重要性(和局限性)。(2) 候选状态:目前的系统和拟议的修改。(3) 在21世纪努力使分类学规模化和系统化。来自四大洲的39位与会者为这第一个系列研讨会做出了贡献,这为以后更有决定性和更有包容性的研讨会奠定了坚实的基础。
第二次研讨会
2018/10
在这个研讨会后,又举行了两次面对面的研讨会。
2018年10月28日至31日期间在美国俄勒冈州胡德河举行的一次研讨会上,24名参会者利用第一期研讨会的民意调查结果,缩小了对微生物系统学的主要问题和可能的解决方案。在全体发言和讨论之后,分组讨论的重点是:(1)更广泛视角下的微生物系统学;(2)目前关于SAG和MAG命名的建议(DNA作为命名类型的一个类别,给予Candidatus名称优先权,建立一个平行的命名系统或不采取行动);(3)生命的基因组树;以及(4)微生物命名—进步主义与保守主义。除了最初的线上研讨会外,此次研讨会还达成了一份共识声明,提出了两条潜在的发展道路:"Plan A",即修正ICNP,允许DNA序列数据作为命名类型的一个类别;或在 "计划A "失败之前,另一个 "Plan B",即开发一个基于DNA序列数据制定新的命名规则,作为已培养和未培养原核生物的统一命名类型。
第三次研讨会
2019/4
第三次研讨会于2019年4月8-9日在美国加州举行,有27人参加。它重点讨论了在前几次研讨会提出的两个计划下与微生物命名法有关的可扩展性和数据库开发。本次研讨会的地点和时间与美国能源部联合基因组研究所(JGI)的 "能源和环境的基因组学会议 "相协调,以利用JGI和与会者中强大的数据库和生物信息学专业知识。
作为研讨会的重点,主要问题如下:(1) 哪些是最紧迫的分类数据库问题,可以帮助将微生物分类学推向下一个十年?(2) 是否有办法达成共识,使共同的命名法或分类法在多个数据库中被平等对待或切实地交叉引用?(3) 是否有办法在未来促进数据丰富的系统学?
这次研讨会,再加上随后对之前关于修改ICNP以包括DNA序列数据作为命名法类型的替代类别建议的反对投票("Plan A"),最终促成了SeqCode("Plan B")第一稿的撰写。
第四次研讨会
2021/2
在原定于2020年在开普敦举行的ISME18会议上,由于COVID19大流行而取消了一次会议和随后的现场研讨会,最后一系列在线研讨会于2021年2月举行(SeqCode研讨会,ISME(https://www.isme-microbes.org))。注:组委会与国际微生物生态学会(ISME)达成协议,在他们的网站上托管SeqCode。
该研讨会围绕着SeqCode的第一份完整的草案,在研讨会前与所有参与者分享,以推动对该文件及其基本原则的批判性审查。最后一个系列研讨会包括两个部分,每个都有两个会议,一个是为方便欧洲、非洲和美洲的参与者而安排的;另一个是为亚洲和大洋洲的参与者安排的。它是由ISME共同主办的,作为SeqCode管理的发展伙伴关系的一部分。第一个部分的主题是 "Path forward for naming the uncultivated",包括六个预先录制的讲座,介绍各种主题和讨论及分组会议。第二个部分的主题是 "Path forward to implementations and adoption of the SeqCode",包括13个关于真核生物系统学、数据库和相关主题的演讲。这一系列研讨会备受期待,来自世界各地的广大微生物学家参加了研讨会,包括来自6大洲42个国家的848名注册者和至少575名与会者。与会者涵盖了微生物学内广泛的分支学科,包括微生物生态学家和系统论者。这两个群体并不经常互动,两个群体的大力参与是研讨会的一个优势,且共有26%的受访者称自己是研究生。我们注意到,尽管有大量的科学家使用分类学名称,但微生物系统学的培训几乎不存在。因此,职业发展是这些研讨会的一个重要成果。共有95%的受访者表示研讨会的内容和成果对他们和/或他们的领域是有用的,90%的受访者表示他们将来可能会使用SeqCode。鉴于SeqCode的强烈参与和近乎一致的支持,SeqCode委员会纳入了分组的反馈,这些分组解决了关于SeqCode的一些关键问题,报告摘要及更多信息详见原文附件。
- 结果 -
在认识到在实施 "Plan B "中进一步的组织参与的重要性后,一个特设的SeqCode组织委员会举办了一系列的在线研讨会(https://www.isme-microbes.org/reports-sponsored-events),正如方法中所述,已吸引了来自6大洲42个国家的广泛的微生物学学科的848名注册者参与。超过90%的参与者表示他们将使用接受DNA序列作为分类的新规则(https://www.isme-microbes.org/ sites/default/files/reports/Path_forward_Naming_Uncultivated.pdf)。鉴于踊跃的参与和近乎一致的支持,SeqCode组委会仔细审议了各个组织/学科的建议并采取了行动,正如方法中所述。成果是编写了SeqCode(正式名称为The Code of Nomenclature of Prokaryotes Described from Sequence Data; 详见补充材料),并在系统上取得了进展以实施它。这些行动启动了一个进程,目标是通过社群的支持和行动来实施SeqCode(Table 1),本文是一个关键但早期的步骤。

SeqCode使用基因组序列数据作为已培养的和未培养的微生物分型的共同“货币”,并遵循类似于ICNP的优先级的规则。从本质上讲,这两种编码的规则都指出,在某一特定位置的分类群的最早有效公布的名称是正确的名称(具有优先权),并遵守历史先例和稳定命名法。SeqCode也承认根据ICNP的规则有效公布的名称的优先权,只要它们不违反根据SeqCode公布的名称的优先权,从而最大限度地减少系统之间的分歧。
通过SeqCode Registry进行名称验证
在SeqCode下验证的分类名称将在SeqCode Registry中捕获,SeqCode Registry是一个注册门户网站,通过该网站注册、验证名称和命名类型并将其链接到元数据。SeqCode Registry的三个主要目标: (1)根据SeqCode进行名称注册和评估; (2)目前文献中使用的候选菌名称的自动识别,以便通过SeqCode的验证,将其中许多名称规范化和标准化; (3)维护在SeqCode下验证的标准的、公开的名称列表,以及关键链接和机器可读的元数据。虽然仍在开发中,但草稿版本目前可在https://seqco.de/上获得。它的所有公共数据都可以访问和再次使用的,除非另有说明,底层代码是根据麻省理工学院许可条款作为开源发布的。完成后,SeqCode Registry将提供用户友好的图形界面访问其相关资源,以及以JavaScript对象符号格式的计算机可读条目,方便第三方服务集成。下文和补充资料提供了使用该系统的例子,供在不同出版情况下登记名称之用。
目前,通过SeqCode注册表有两种不同的注册和验证名称的机制(Fig. 1); 第三种机制可能在未来出现。在最好的情况下,数据将在发表前通过预注册过程输入和审查,该过程发生在初次提交或重新提交稿件之前(Fig. 1,左侧蓝色箭头,路径1)。
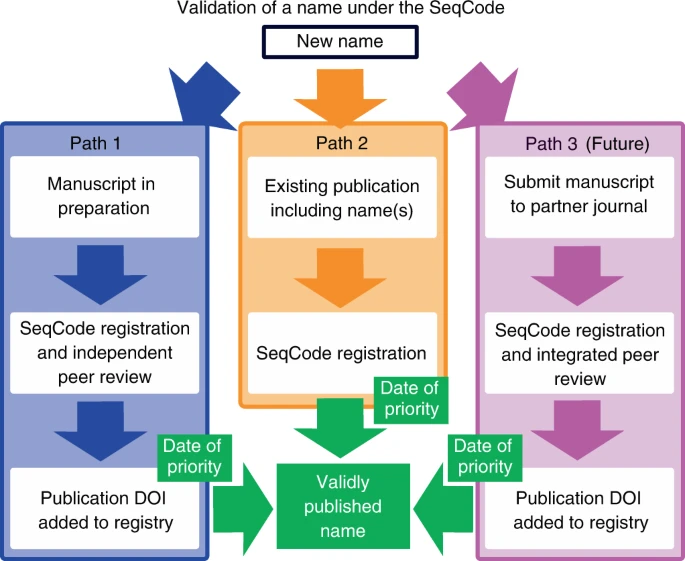
图1. SeqCode下名称的验证过程
(目前有两种机制,未来可能会有第三种。在推荐的机制中(蓝色箭头,路径1),名称和元数据进入SeqCode Registry的草案注册与有效发布的准备同时进行。在注册中心内,数据质量和名称同义词检查以及管理员审查如表2和表3所述,导致临时接受符合SeqCode规则的提案。此程序可确保数据质量,并避免发表后要求更正的勘误表。将出版物的DOI输入书记官处标志着优先的时间和日期。因为SeqCode要求使用分类单元的最早名称,所以优先级日期将此名称的优先级确定为分类单元的唯一有效名称。第二个(橙色箭头,路径2) 用于已经发布的名称,如Candidatus名称。名称和元数据被输入到Registry中。自动检查和SeqCode管理员审查合合性和接受建议完成注册并标记优先时间和日期。此时,可以删除Candidatus指定。第三种机制可以在未来与一个或多个期刊合作发展(粉色箭头,路径3)。它将包括同行审查和登记处馆长审查,作为验证拟议名称的综合途径。被接受论文的DOI的发布标志着优先的时间和日期。关于通过路径1和2注册的具体例子,请参阅正文和补充信息。
该路径允许SeqCode Registry执行自动检查并提供管理器输入,这两者都可以作为指导用户社区的资源。通过提供这些预检查,路径1发挥了以下两个重要作用。(1)预注册时的自动检查和管理员输入,可以防止名称在发表前出现同义词或拉丁化问题等错误,从而防止发表后名称更改造成的混乱。因此,这个过程有点类似于国际系统与进化微生物学杂志(IJSEM )同行评审时的手动命名检查。通过尽可能地自动化该过程,目标是最大限度地提高速度和灵活性,并最小化人为错误。同样,数据质量检查通过确保作为命名自然类型的基因组数据具有足够的质量来指导用户社群。目前,基因组质量和完整性数据由用户输入,并根据需求和建议进行检查,尽管在未来这些检查将是自动化的。(2)预注册期间生成的SeqCode标识符统一资源定位符(URLs)可以包含在作为有效出版物(建议使用新名称的出版物)提交的手稿中。
这些URLs允许同行评审员和编辑访问预注册的名称,以确保它们通过SeqCode检查。这个过程应该改善和简化同行对新名称和相关基因组的审查,因为SeqCode Registry在预注册阶段的批准可以使人们相信这些名称不存在同义词和不良拉丁化等问题,而且作为命名类型的序列数据具有足够的质量。还应该注意的是,任何人在任何时候都可以在SeqCode Registry处提出根据SeqCode验证的名称的小写正体的变化(minor orthographic variants),而不需要公布勘误表。,这也是为了尽量减少工作量和避免混淆。关于这些正字变体的决定将由策展人进行裁判。在路径1下,注册程序的完成以及名称的优先权日期是数字对象标识符(DOI)被输入SeqCode Registry的日期。这通常是由作者完成的,但是,如果SeqCode标识符URLs被用于有效的出版物中,那么一旦稿件出版,DOI将被SeqCode Registry自动捕获,完成注册过程。
第二种机制(中间或橙色箭头,路径2)允许注册和验证已经发表的名字,包括Candidatus名字。名称和元数据被输入到注册中心,并通过路径1下实施的相同的自动检查进行筛选。然后,SeqCode的管理员对名称进行审查,接受条目完成注册并标记名称的优先权日期。在这一点上,该名称是有效的,Candidatus的名称可以被移除。我们注意到,路径2不如路径1,因为命名法或基因组质量的问题在名称公布前不会被标记和纠正。因此,根据SeqCode,在文献中发表的名称最终可能被修改或无效;然而,如上所述,SeqCode和注册中心的设计尽可能灵活,从而更好地服务于社群。这是有可能的因为SeqCode Registry同时是注册和验证系统以及根据SeqCode验证的官方和最新的名称列表。
第三种机制(右侧或粉色箭头,路径3)将涉及到同时进行的同行评审和注册处管理人员的评审,作为验证所提议的名称的综合路径,类似于IJSEM的综合评审系统,它为ICNP下提议的名称服务。
在发表前通过路径 1 应用 SeqCode
下面将以WolframiiRaptor gerlachensis和相关的分类群为例,并在补充材料中说明这一流程如何发挥作用的具体例子。简言之,SeqCode的几位作者(M.P.,A.-L.R.和B.H.)最近完成了对基因组分类数据库(GTDB)家族中以前未被描述的一组古细菌的联合培养/宏基因组学研究,该家族被指定为Thermoproteota门中的NZ13-MGT,在以前的文献中也讨论过,被称为'Aigarchaeota' groups 4, 5 and 7 。该研究最初关注来自美国内华达州大沸泉沉积物的厌氧富集培养物,其中含有该分类群的一个成员,该成员需要钨才能在发酵条件下的玉米秸秆或混合糖上生长。荧光原位杂交结合纳米级二次离子质谱法被用来确认木糖是首选底物。该类群由一个高质量的宏基因组组装基因组(MAG)代表,尽管该 MAG 与来自相同富集培养物及其所在沉积物的不同样品的质量较低的 MAG 形成了 >99.5% 的平均核苷酸同一性 (ANI) 簇。为了扩大研究范围,从其他陆地和海洋热液系统的宏基因组中组装了另外77个由GTDB-Tk13分配给GTDB家族NZ13-MGT的高质量MAGs。
Table 2中建议根据SeqCode命名的物种或亚种包括一个以上的基因组,这与ICNP的一般建议相类似。即为新的分类学名称的建议提供多个菌株的特征,这对MAG和单扩增基因组(SAG)特别重要,因为我们在准确的宏基因组数据分箱上存在挑战,而且大多数SAG的完整性不高。

本研究使用FastANI14将78个高质量的MAGs去复制为11个>95% ANI簇(物种簇),并对连接的标记基因集进行系统发育分析,确认每个ANI簇都是单系的。共有9个物种簇由2-16个来自不同采样日期和/或地泉的宏基因组的高质量MAGs所代表。比较每个物种簇的多个MAGs,可以评估:(1)使用多个标记基因集,每个物种的单系性; (2)通常单拷贝的保守标记基因的多个副本真实存在,用于评估基因组完整性和污染的保守标记基因真实不存在; (3)编码重要功能的基因(在本例中为钨酸盐转运蛋白、钨酶和与能量守恒相关的基因)同源物的存在; (4)共有基因含量; (5)同一物种内基因组的含量和大小相似。这些比较加强了关于所提出的能量守恒模式、生物体及其酶系统进化的结论,并允许对有问题的MAGs进行识别和排除。我们注意到,从几个GTDB物种代表中检测和系统发育分析了MAGs,但大多数MAGs的质量不足以在SeqCode中命名(Table 3)。

最后,有11个物种群的类型序列符合SeqCode的数据质量标准(Table 3),从而提出了11个物种名称以及它们在SeqCode下的母类群。名称是按照ICNP附录9的一般建议和其他指南的拉丁文规则形成的,并由命名专家A.Oren检查。在未来,它们将由SeqCode Registry的管理员审核。在补充材料“SeqCode preregistration”部分详细描述了预注册的过程。在预注册之后,有效出版物将被提交给同行评审。有效出版物包括每个分类名称的以下内容:(1)明确指定命名类型;(2)指定分类等级;以及(3)新名称的词源(Table 2)。命名建议是在原始资料中提出的,下面是两个分类等级的例子。虽然在SeqCode中不要求原生境,但它们对于分类学描述是有用的,因为它们将关键信息汇编在一起。表格也可以使用,其例子在补充资料中。我们在此注意到,一旦预注册完成,SeqCode Registry就会产生原始资料(protologue)。这些资料对科学界是有用的,因为它们可以通过出版物中的URL链接修改后作为出版物中的资料,或在注册中心内随时在线访问。
在有效的出版物中,我们建议为先前未描述的WolframiiRaptoraceae科进行命名,这是先前未描述的WolframiiRaptor属的母分类单元。这个科的名称取代了GTDB中的名称NZ13-MGT,并在补充材料中做了说明。下面的例子描述了以前未描述的Wolframiiraptor属。请注意,对于一个属来说,命名类型是一个种,就像在ICNP中一样。括号中的说明解释了与SeqCode的原则、规则和建议有关的原始资料。
Wolframiiraptor (Wolf.ra.mi.i.rap’tor N.L. neut. N. wolframium, tungsten; L. masc. n. raptor, snatcher or thief; N.L. masc. n.Wolframiiraptor, snatcher of tungsten)。因此,Wolframiiraptor中文可理解为“掠夺/偷钨元素的人”。本文根据SeqCode规则26.4和26.5指定分类等级(属)和词源。
本属成员已从美国大盆地和黄石国家公园的地热泉,以及中国腾冲境内被发现。代表属内不同物种的基因组的平均氨基酸同源性(AAI)值在81%到90%之间。根据祖先状态重建分析,编码细胞色素c氧化酶亚基、好氧一氧化碳脱氢酶大亚基和硫化物: 醌氧化还原酶(Sqr)的基因可能丢失,表明该属成员可能是严格厌氧菌,不能硫化物氧化。该属基因组编码钨酸(Tup) ABC转运蛋白的一个tupA亚基,包含多个编码钨依赖性氧化还原酶的基因,包括三种推定的醛类: 铁还蛋白氧化还原酶(AOR),一种甲醛:铁还蛋白氧化还原酶(FOR-like)和一种甘油醛-3-磷酸: 铁还蛋白氧化还原酶(GAPOR)类蛋白。该分类单元被系统基因组学、AAI和相对进化差异支持其为属水平的类群。(本文包括该分类单元的描述,参见建议26。这类文本是推荐的,但在SeqCode中不是必需的。)。更多示例详见原文。
通过路径2将SeqCode应用于已经公布的名称
除了已经公布的名称外,还包括Candidatus名称。SeqCode也允许注册以前公布的名称,如符合其规则的候选名称。Candidatus是一个在命名法中缺乏优先权和地位的临时状态,被归入ICNP的非合法性附录11中。它是为那些 "不仅仅是有核酸序列 "的生物体开发的。自其诞生以来,人们一直建议在自然样品中对该类群进行可视化处理(在做图时考虑进来),但这很少有人这样做。有人认为,在ICNP下,Candidatus名称应该被赋予优先权;然而,这一建议也被ICSP否决了。许多Candidatus名称可能被证明是短暂的,在SeqCode下对这些名称进行验证将给予它们优先权,并且可以放弃对Candidatus的命名(Fig.1,路径2)。这些名称具有特殊的重要性,因为目前已经编制了一个超过1000个Candidatus名称的目录,最近作为鸡粪便微生物组研究的一部分发表了917个Candidatus名称。SeqCode在有效的出版物中特意制定了很少的要求,以允许这些和其他名称得到验证(Table 2)。事实上,只要分类群在有效出版物中被命名,并且基因组符合命名类型所要求的数据质量标准,文献中的任何Candidatus名称都可以在路径2下被验证(例子见补充信息)。这是有可能的,因为关键的数据包括命名类型的指定,可以在验证期间在SeqCode注册表中获取。我们计划启动这项工作,这将与社群合作完成。
然而,我们欢迎Candidatus类群的作者自己来验证那些已经有效发表并符合序列质量标准的名称。由于SeqCode注册中心已经开始运作,这项工作可以立即开始。验证大量Candidatus名称的基本程序是:(1)评估分配给每个Candidatus分类群的基因组序列的数据质量;(2)如果一个序列的质量足以作为一个类型,联系作者检查由SeqCode Registry生成的自动填充模板,并完成缺失的数据字段;(3)在SeqCode Registry完成验证;(4)与社群的合作者发表一篇论文,宣布对名称的验证。这个项目将完成Candidatus名称的验证,集中这些分类群的名称和元数据,起到一个重要的扩展功能,引导社区关于SeqCode的原则和实施,并为社群提供一个反馈的渠道。
数据标准
Table 3总结了SeqCode组委会关于数据和报告要求的最低标准的建议。选择这些标准是为了精确地描述物种,并纳入了基因组标准协会的许多建议。SeqCode组织委员会详细讨论了使用DNA序列作为类型的新名称的原始发布标准。大多数人认为,发表要求应能够命名所有科学上得到充分支持的名称。例如,没有必要在出版物中要求基因组登录号,因为它可以在SeqCode Registry中随时获得。这将允许在尚未明确识别类型基因组的情况下对Candidatus菌名进行临时注册。但是,我们强烈建议今后的出版物应载有登记号。同样,讨论了是否需要或推荐16S核糖体RNA序列。大多数人的意见是,16S rRNA序列不是物种鉴定的必要条件,不应该被要求。尽管如此,整个委员会都认识到,原核生物的现代分类学是基于16S rRNA的系统发育,包含一个精确的16S rRNA序列提供了该分类学以及一个巨大的环境核型数据库的访问。由于这些原因,我们强烈建议包含一个精确的16S rRNA序列,尽管我们认识到rRNA基因可能很难组装和准确分箱,因为它们经常出现在多个拷贝中,不符合编码序列的核苷酸词频模式。虽然在规则本身外,这些标准是SeqCode的附录,但通常应该应用这些标准,除非有强有力的理由验证具有较低质量基因组的名称作为类型(例如包含生理学、生态学或进化方面的大型数据集的中等质量的基因组)。我们期望这些标准将随着社群反馈和方法改进而不断发展。虽然SeqCode本身很全面,但我们还开发了一些资源来指导社区,包括用于命名的数据类型的术语表和示例(详见补充信息)。
- 讨论 -
SeqCode的一个目标是扭转分类学名称在主要文献中发表但没有有效发表的趋势。尽管许多社群可以自由发表不符合命名法则的分类学名称,但我们认为命名法则和分类学框架通过促进客观性、最佳实践、交流和数据互操作性为更大的社区服务。然而,ICNP对可行的和可获得的类型菌株的独特限制疏远了许多微生物学家,这迫使他们在ICNP规定之外发表物种命名。SeqCode通过提供一个有效的和用户友好的资源来解决这个问题,以满足更广泛的研究团体的共同利益。SeqCode包含了可查找性、可访问性、互操作性和可重用性(FAIR)的原则,注册表是用可交互操作的数据结构开发的,以促进SeqCode名称在微生物学和更广泛的生物学研究界的全球生物多样性目录中的共享(例如NCBI、GTDB、MiGA、LPSN、生命目录和全球生物多样性信息机构)。
最后,我们强调几个重要的问题。首先,SeqCode的目的不是要阻碍培养。混合或纯培养物的培养可以在受控条件下测试从基因组预测的特性。此外,我们强烈鼓励研究者将菌株保存在培养物中,以提高菌株的可用性,使表型性状的可重复性得到评估,为生物化学和生物技术提供资源并促进国际合作。第二,像所有其他的命名法则一样,SeqCode并没有提供关于分类群划分的规则或建议。现有的和改进的方法和数据结构可用于这一目的,关于描述以前未描述的类群的建议必须通过同行评审来解决。最后,这是SeqCode的第一个版本,我们希望它能随着社群对系统的进一步开发而不断发展。由于我们希望为广大的微生物学研究界服务,我们将让社区参与进来,收集反馈意见并制定SeqCode的管理细则。这个规则是由自下而上的愿景驱动的,以改善整个微生物科学的交流。因此,我们认为这个 "SeqCode v.1.0 "是迈向一个统一的命名系统的必要的第一步,以沟通原核生物的全部多样性,我们将与社群合作以实现这个愿景。
参考文献
Hedlund, B.P., Chuvochina, M., Hugenholtz, P. et al. SeqCode: a nomenclatural code for prokaryotes described from sequence data. Nat Microbiol (2022). https://doi.org/10.1038/s41564-022-01214-9
- 第一作者简介 -

美国内华达大学
Brian P. Hedlund
博士,研究员
Hedlund于2000年在华盛顿大学获得博士学位,并在Jim Staley的实验室从事多环芳烃降解海洋细菌的培养和特征研究以及Verrucomicrobiota的生物学研究。他在德国雷根斯堡大学的Karl Stetter那里完成了博士后培训并开始研究嗜热菌,从2003年开始,他在内华达大学拉斯维加斯分校作为独立研究员继续研究。Hedlund的研究重点是地热生态系统的微生物学和生物地球化学,“微生物暗物质”的基因组探索,以及肠道微生物组在预防艰难梭菌感染中的作用。Hedlund博士是Antonie van Leeuwenhoek杂志的编辑,Bergey’s Manual Trust的成员,也是微生物分类学权威参考手册Bergey’s Manual of Systematics of Archaea and Bacteria的编辑。
- 通讯作者简介 -

南非比勒陀利亚大学
Stephanus N. Venter
博士,研究员
Venter是南非比勒陀利亚大学微生物学和植物病理学系主任。他的主要研究兴趣是细菌进化、系统学和多样性,尤其关注与如何应用细菌学方面的知识来理解细菌和细菌群落在不同环境中的功能特性。Venter及其研究小组在解决这些问题时主要侧重于使用分子、测序和基因组学等方法,目前他所关注的两个主要领域是与植物和水生环境相关的细菌。

美国佐治亚大学
William B. Whitman
院士,研究员
Whitman是美国佐治亚大学微生物系系主任,兼任International Journal of Systematic Bacte riology副主编,JournalofBacteriology编委,Bergeys Manual责任主编,美国NIH,DOE,NSF等项目评审专家。并曾担任多个重要微生物国际会议的主席,在国际微生物学领域具有很高的学术地位和声誉。Whitman教授长期专注于微生物学研究,尤其在产甲烷菌的研究方面取得了杰出的研究成果,研究成果在Science、Nature、PNAS、JBC等国际顶级期刊上发表。由于在微生物研究领域的突出贡献,Whitman博士被遴选为美国微生物科学院(American Academy of Microbiology AAM)院士。美国科学促进会会士(The American Association for the Advancement of Science Fellow),曾荣获美国青年科学家总统奖,伯杰氏奖章等多个杰出奖项。
猜你喜欢
iMeta简介 高引文章 高颜值绘图imageGP 网络分析iNAP
iMeta网页工具 代谢组MetOrigin 美吉云乳酸化预测DeepKla
iMeta综述 肠菌菌群 植物菌群 口腔菌群 蛋白质结构预测
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature
一文读懂:宏基因组 寄生虫益处 进化树 必备技能:提问 搜索 Endnote
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流快速解决科研困难,我们建立了“宏基因组”讨论群,己有国内外6000+ 科研人员加入。请添加主编微信meta-genomics带你入群,务必备注“姓名-单位-研究方向-职称/年级”。高级职称请注明身份,另有海内外微生物PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。
点击阅读原文


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









