






二代测序 (NGS) 的发展已经彻底改变了许多领域的研究。在很多临床研究队列研究中,微生物甚至被誉为人类的第二基因组,参与多种疾病的生理及病理生理机制。然而,海量的测序数据以及各种统计分析及模型算法的引入,显著增加微生物数据分析研究初学者的学习难度。特别是在研究初期需要频繁的进行数据及代码操作时,繁杂的数据分析代码极大的提高了各种意想不到错误的风险性。因此,需要一种高效便捷的微生物组数据分析工具能够协助研究人员快速处理微生物下游数据。 EasyMicroPlot 软件包旨在集成目前大部分主流的微生物数据分析及可视化模块,为微生物研究初学者提供一个基于R平台的便捷分析工具。
### 1.基本特点
EasyMicroPlot 软件包(简称:EMP)主要面向低代码经验的微生物数据分析人员,尽可能优化很多繁杂的操作。只需要**表型数据**、**微生物丰度表**和**样本分组表**即可快速完成目前主流的微生物下游数据分析工作。
具有以下特点:
* 1. EMP包可以直接识别不同级别的微生物数据,无须手动指定修改数据文件名
* 2. EMP包计算中进行样本重组和剔除样本时,只需修改mapping文件,无须修改微生物和表型数据。
* 3. EMP包的大部分微生物计算模块相互独立,可以根据实验需要手动选择
* 4. EMP包提供了一种基于最小相对丰度和物种组内出现率的核心微生物的过滤方法
* 5. EMP包大部分图示支持交互式图形,便于查询更多信息内容
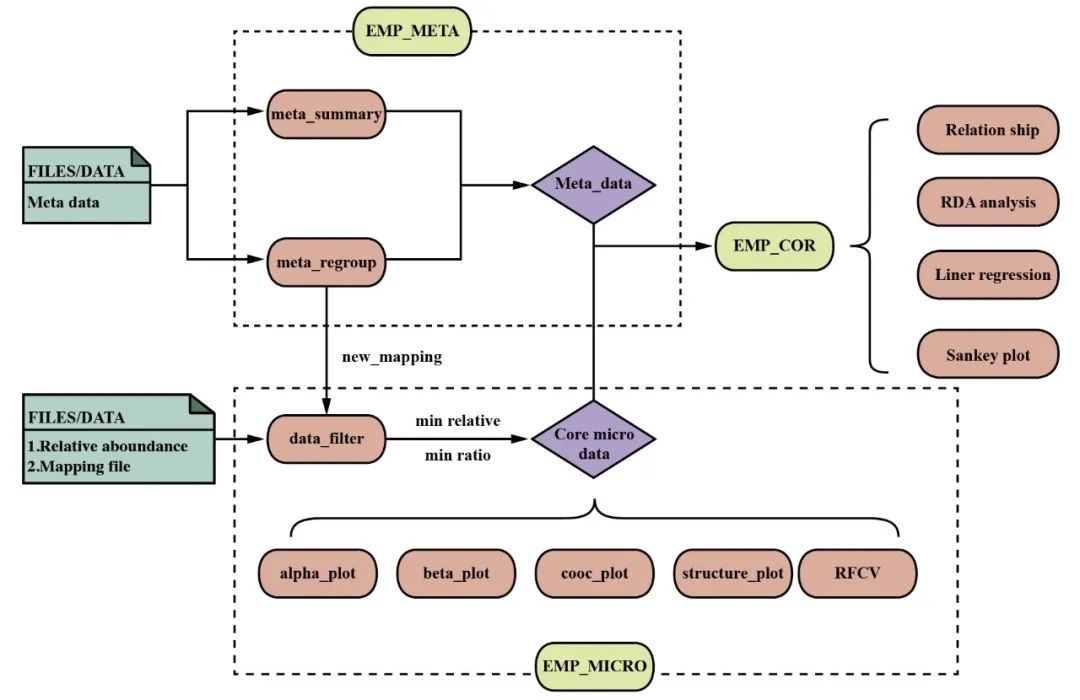
overview.png
### 2. 安装方法及教程地址
**Github安装方法**
```R
install.packages('remotes')
remotes::install_github("https://github.com/xielab2017/EasyMicroPlot",subdir='Version_0.5')
```
**Gitee安装方法**
考虑到github的网络连接问题,EMP包提供了中国境内Gitee安装方式。
首先要确保电脑内已经安装Git工具
Git工具官方下载安装地址:https://git-scm.com/downloads (首次安装Git后,需重启R或者电脑)
```R
install.packages('remotes')
remotes::install_git("https://gitee.com/xielab2017/EasyMicroPlot/",subdir='Version_0.5')
```
EMP包全部功能详细中文教程:https://xielab2017.github.io/EasyMicroPlot_tutorial/
EMP包BUG提交及讨论QQ群:729506293
### 3. 分析前准备工作
首先将满足格式要求的文件放在R工作的本地文件夹内,再`library(EasyMicroPlot)`加载EMP包。
具体模块功能可以在Rstudio中输入`help (EMP) `查看R内置说明及示例文档
示例数据下载地址:
https://github.com/xielab2017/EasyMicroPlot/tree/main/Demo%20data
### 4 快速分析模式
此模式能够帮助用户在首次接触项目数据时,快速批量进行一系列主流的微生物分析,了解微生物数据的大致情况。
**单次模式**
此模式下能够快速根据mapping文档从微生物数据集中抽取数据,批量进行主流的核心微生物过滤、多样性分析、共发生网络分析、随机森林分析、结构组成分析等。
```R
library(EasyMicroPlot) # 加载包
# 简要模式
EMP_MICRO(dir = '16s_data/',design = 'mapping/mapping.txt',
min_relative = 0.001,min_ratio = 0.7,pattern = 'txt')
```
**批量模式**
用户可以根据实验需要,在一个mapping文件下设计不同分组或者不同样本的多个mapping文件,批量进行下游分析计算
Tip: 熟悉R语言的用户,也可以设计不同过滤条件、不同mapping分组快速完成初步分析。
```R
# 批量执行多个分组
mapping_files <- c('mapping1.txt','mapping2.txt')
for (i in mapping_files) {
mapping <- paste0('mapping/',i)
EMP_MICRO(dir = '16s_data/',design = mapping,
min_relative = 0.001,min_ratio = 0.7,pattern = 'txt',
distance = c('bray','jaccard','euclidean'),
output_folder = paste0('Result/',strsplit(i,'.txt')[[1]]))
}
```
**输出结果文件说明**
```R
Result # 输出结果文件名
│
├── alpha_result # 微生物各级别alpha多样性结果汇总
│ ├── Post-Hoc # onewayANOVA事后多重检验详细结果
│ ├── data # alpha多样性值
│ ├── pic # alpha多样性图形结果
│ └── html # alpha多样性可交互式图形结果
├── beta_result # beta多样性结果汇总
│ ├── bray_0.001_0.7 # 基于Bray Curtis距离计算的结果
│ └── jaccard_0.001_0.7# 基于Binary Jaccard距离计算的结果
├── cooc_result # 微生物各级别共发生网络结果汇总
│ ├── cooc_info # 微生物各级别各组节点相关性系数及检验结果
│ ├── net_profile # 微生物各级别共发生网络基本属性结果
│ ├── network # 微生物各级别各组共发生网络基本图形结果
│ └── vertex # 微生物各级别各组共发生网络节点重要性评估结果
├── core_data # 微生物各级别核心微生物结果汇总
│ ├── class_0.001_70%.txt
│ ├── class_0.001_70%_info.txt
│ ├── family_0.001_70%.txt
│ ├── family_0.001_70%_info.txt
│ ├── genus_0.001_70%.txt
│ ├── genus_0.001_70%_info.txt
│ ├── order_0.001_70%.txt
│ ├── order_0.001_70%_info.txt
│ ├── phylum_0.001_70%.txt
│ ├── phylum_0.001_70%_info.txt
│ ├── species_0.001_70%.txt
│ └── species_0.001_70%_info.txt
└── structure_result #微生物各级别物种结构结果汇总
│ ├── pic # 基本结构组成图
│ ├── taxonomy # 物种详细注释
│ └── top_abundance # Top物种结果
├── RFCV_result #微生物种级别随机森林模型结果汇总,如需采用其他级别,请修改RFCV_estimate参数
│ ├── Imprortance # 不同随机数种子下随机森林物种重要性评估图
│ ├── model # 随机森林交叉验证错误率曲线图
└── └── taxonomy # 随机森林交叉验证筛选物种统计及可视化结果图
```
### 5 精细分析模式
此模式能够帮助用户针对性的完成各种主流微生物下游数据分析,更多详细功能可以参考完整教程。
### 5.1 核心微生物过滤
在微生物注释分析结果中,可以发现存在相当多的”稀有物种“。这些”稀有物种“具备相对丰度或者物种组内出现率低的特点,对于筛选组间微生物差异性造成了极强的干扰。特别是筛选关键微生物的分析中,机器学习算法,例如随机森林、LEFse,很容易将这些个别组内存在的稀有物种,识别为潜在标志物种,因此有必要在正式分析前将这些稀有物种根据统一的标准进行过滤。本章节的`data_filter`模块,根据`物种相对丰度`和`物种组内出现率`对物种进行过滤,并筛选出不同注释级别的核心微生物。过滤的基本流程为:首先将不同注释级别中将全部出现的物种进行编号,再根据设定的`最小物种相对丰度`使低于此阈值的丰度转换为0,最后将要求核心物种必须满足在至少一个分组内`物种组内出现率`高于预先设定的阈值,其余物种则判断为”稀有物种“进行过滤。
**提示**:如果用户不需要进行这种核心微生物的过滤方法,只需将`min_relative`和` min_ratio `均设置为0即可。本文示例的`min_relative=0.001`和` min_ratio=0.7 `仅供参考,即意味着核心微生物的最小相对丰度必须大于1‰,且在至少一个分组内出现率超过70%。
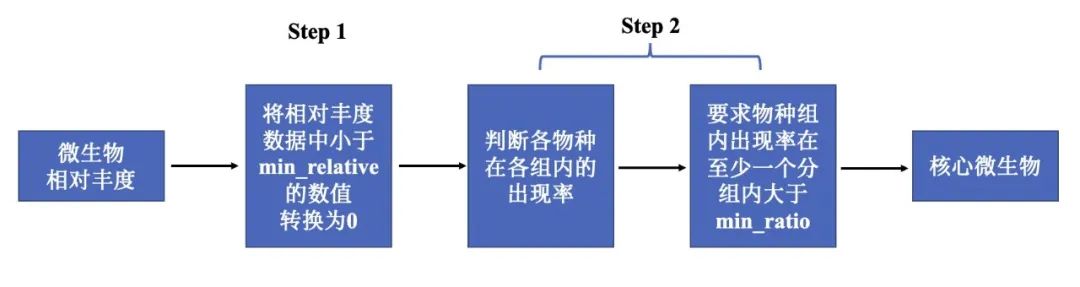
data_filter.png
```R
library(EasyMicroPlot) # 加载包
core_data <- data_filter(dir = '16s_data/',design = 'mapping/mapping.txt',
min_relative = 0.001,min_ratio = 0.7,pattern='txt')
# 核心物种(门级别)
core_phylum <- core_data$filter_data$phylum
core_phylum <- core_data$filter_data$phylum_ID
# 核心物种(种级别)
core_species <- core_data$filter_data$species
core_species_ID
<- core_data$filter_data$species_ID
```
### 5.2 Alpha多样性
α多样性是微生物下游分析中常见的分析方法,主要用于评估样本组间的物种丰富度。本章节`alpha_plot`模块基于相对丰度数据,内置了4种常见的α多样性的方法,并提供了常规的统计分析和可视化功能。
```R
library(EasyMicroPlot) # 加载包
alpha_re <- alpha_plot(dir = '16s_data/',design = 'mapping/mapping.txt',min_relative = 0.001,min_ratio = 0.7, method = 'ttest') # 这里method也可以选择LSD、SNK等one way anova的统计方式
## alpha_re$result$filter_data 这里存储了前置data_filter函数过滤的核心微生物结果
## alpha_re$result$alpha_result 这里存储了核心微生物各个级别alpha多样性的计算结果
## alpha_re$plot 这里存储了核心微生物各个级别alpha多样性的图形结果
alpha_re$plot$species$pic$Total ## 以种级别为例,此为种级别alpha多样性总图
alpha_re$plot$species$html$Total ## 以种级别为例,此为种级别alpha多样性总图的交互式版本
```
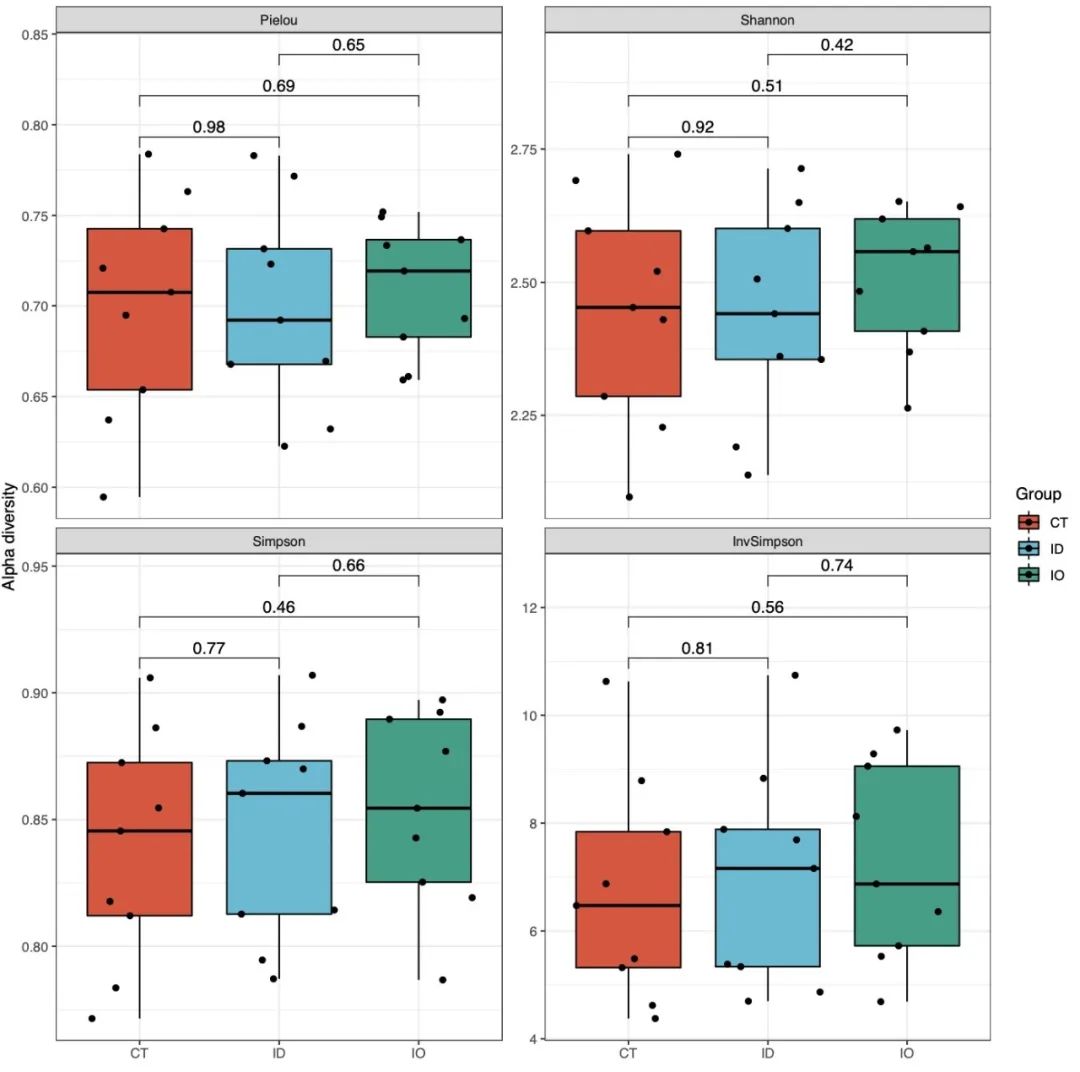
alpha_diversity1.png
### 5.3 Beta多样性
β多样性常用来评估组间微生物物种结构的总体差异性。本章节`beta_plot`模块提供了多种距离算法用于进行维度差异统计及可视化结果。
```R
library(EasyMicroPlot) # 加载包
# 这里method可以选择LSD、SNK等one way anova的统计方式,distance可以选择bray、jaccard、euclidean、gower等距离
beta_re <- beta_plot(dir = '16s_data/',design = 'mapping/mapping.txt',
min_relative = 0.001,min_ratio = 0.7,
distance = 'bray',method = 'ttest')
## beta_re$result$filter_data 这里存储了前置data_filter函数过滤的核心微生物结果
## beta_re$plot 这里存储了核心微生物各个级别beta多样性的图形结果
beta_re$plot$species$pic$p12 ## 以种级别为例,此为种级别beta多样性前两轴的二维平面
beta_re$plot$species$html$p12 ## 此为种级别beta多样性前两轴的二维平面的html版本
## 注意此图形为拼接图形,如果在Rstudio中显示效果不佳,可以将其图形输出,或者直接查看html版本。
## 输出方法为ggplot2标准方法
library(ggplot2)
ggsave(beta_re$plot$species$pic$p12,filename = 'p12.pdf',width=15,height=15)
```
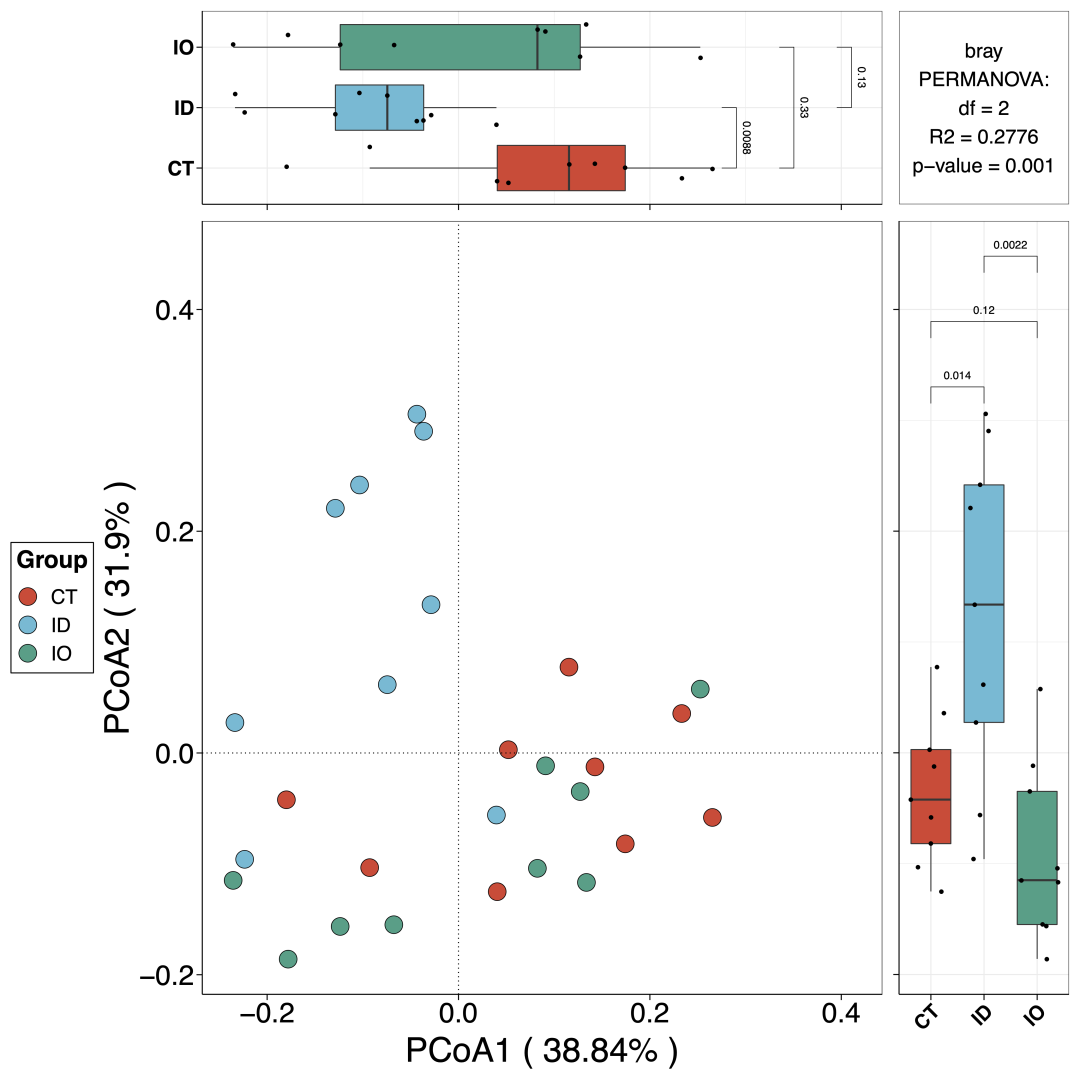
beta_diversity2.png
### 5.4 Co-oc共发生网络
共发生网络分析是利用相关性检验评估微生物物种之间的竞争或者抑制关系,也可以利用图论相关技术来从整体评估微生物物种生态网络的基本属性,筛选可能存在的微生物社区网络及网络关键微生物。`cooc_plot`函数提供了一种基于线性相关性分析计算网络的方式,并内置了三种节点重要性评估的算法,便于发现网络中重要的节点。
```R
library(EasyMicroPlot) # 加载包
# 当group_combie = T 时,此函数将全部分组数据合并为一组进行计算
# 当clust = T 时,将采用贪婪算法识别出网络中的sub-community
# cooc_r 为判定节点之间相关性绝对值的最小阈值,低于此阈值的边将会被过滤
# cooc_p 为判定节点之间相关性绝对值的统计眼见最小值,p大于此值时的边将会被过滤
cooc_re <- cooc_plot(dir = '16s_data/',design = 'mapping/mapping.txt',
min_relative = 0.001,min_ratio = 0.7,group_combie = F,clust = F,
cooc_method = 'spearman',cooc_output = T,
cooc_p = 0.05,cooc_r = 0.3)
##cooc_re$result$filter_data 这里存储了前置data_filter函数过滤的核心微生物结果
##cooc_re$cooc_profile 这里存储了核心微生物各个级别共发生网络的基本属性结果
##cooc_re$plot 这里存储了核心微生物各个级别共发生网络的igraph相关图形信息
```
**注意1**:由于EMP包的共发生网络计算是由igraph包进行计算,暂无法支持ggplot2绘图方式,因此提供了`cooc_output = T`参数,可以将全部图形输出到本地工作目录直接查看。
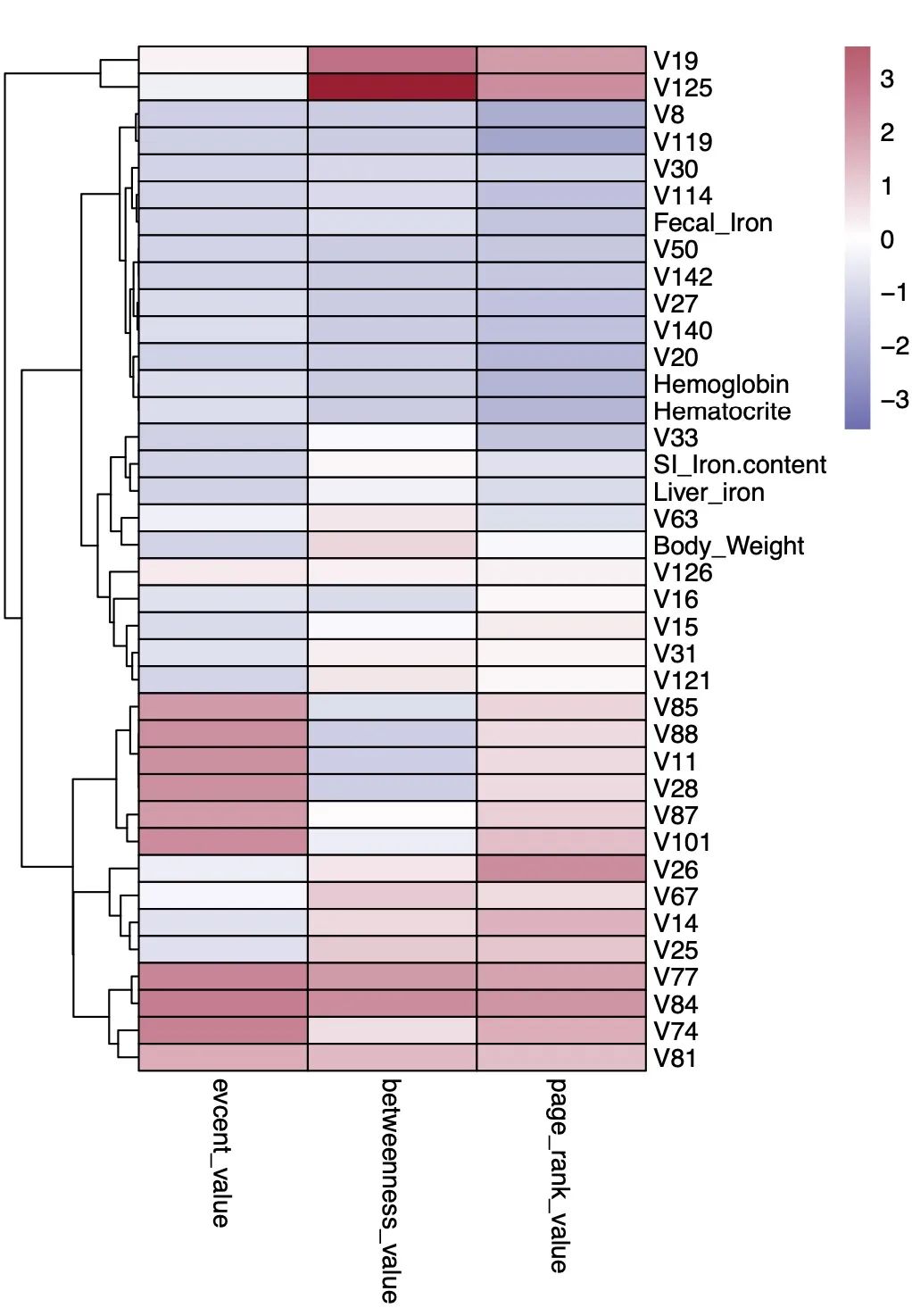
**注意2**:此模块内置了三种图形评估节点重要性的算法`event_value`,`betweenness_value`和`page_rank_value`,并用热图展示。
**注意3**:由于此模块存在着核心微生物和相关性双重过滤,当出现空集时会影响中断结果输出。此时建议减小过滤强度,或者仅在物种较多的级别,如属、种级上进行分析。
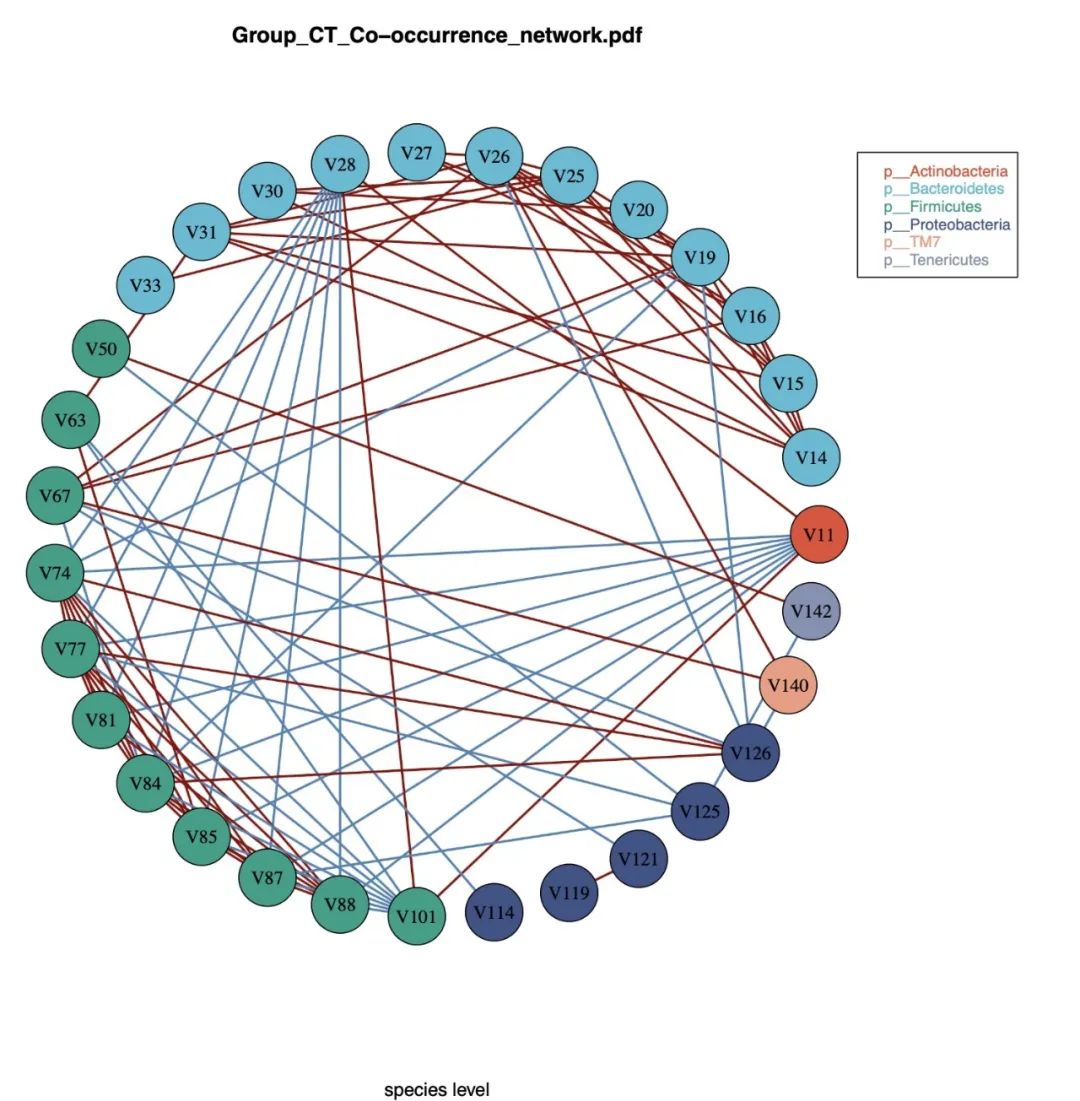
cooc1.png
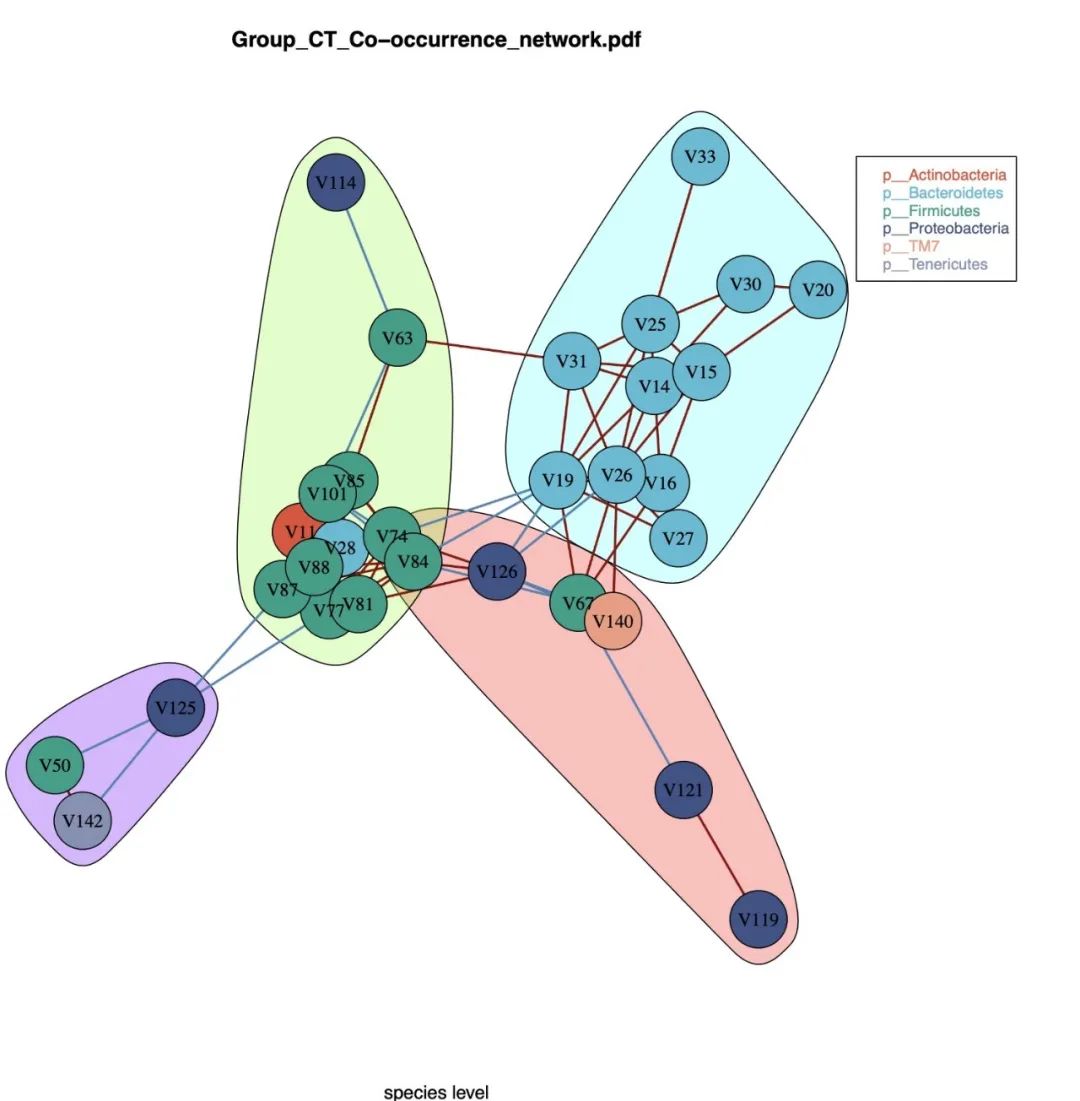
cooc3.png
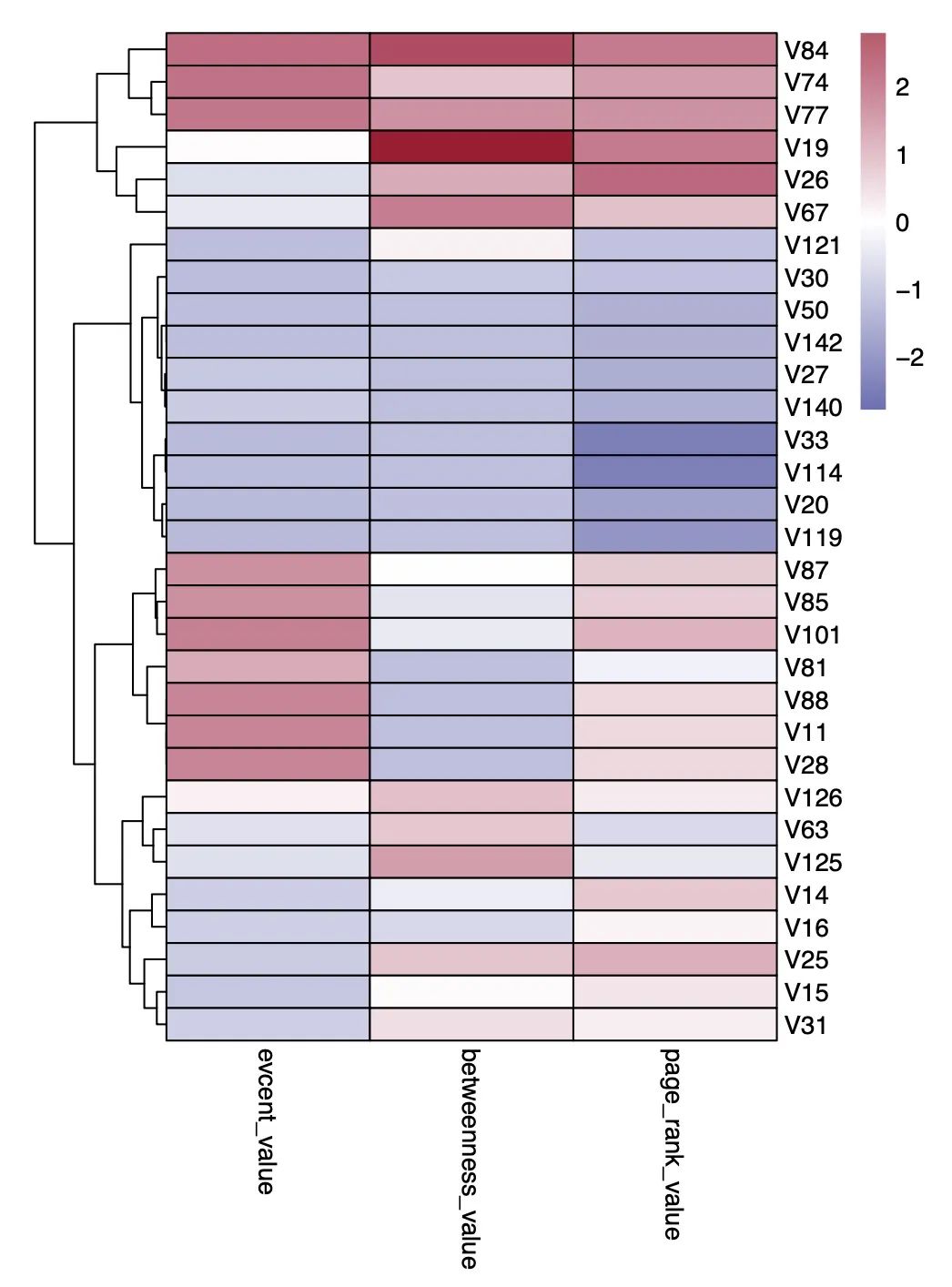
cooc2.png
此外,EMP的`cooc_plot`模块支持将表型数据与微生物数据共同计算共发生网络,便于用户直观的发现表型数据与微生物数据之间的相互关系。
```R
# 整合表型数据联合分析
meta_data <- EMP$iron # 注意数据要符合详细网页教程3.2的格式要求,这里是内置的示例数据
cooc_re <- cooc_plot(dir = '16s_data/',design = 'mapping/mapping.txt',meta = meta_data,
min_relative = 0.001,min_ratio = 0.7,cooc_method = 'spearman',cooc_output = T,
cooc_p = 0.05,cooc_r = 0.3,clust = T )
```
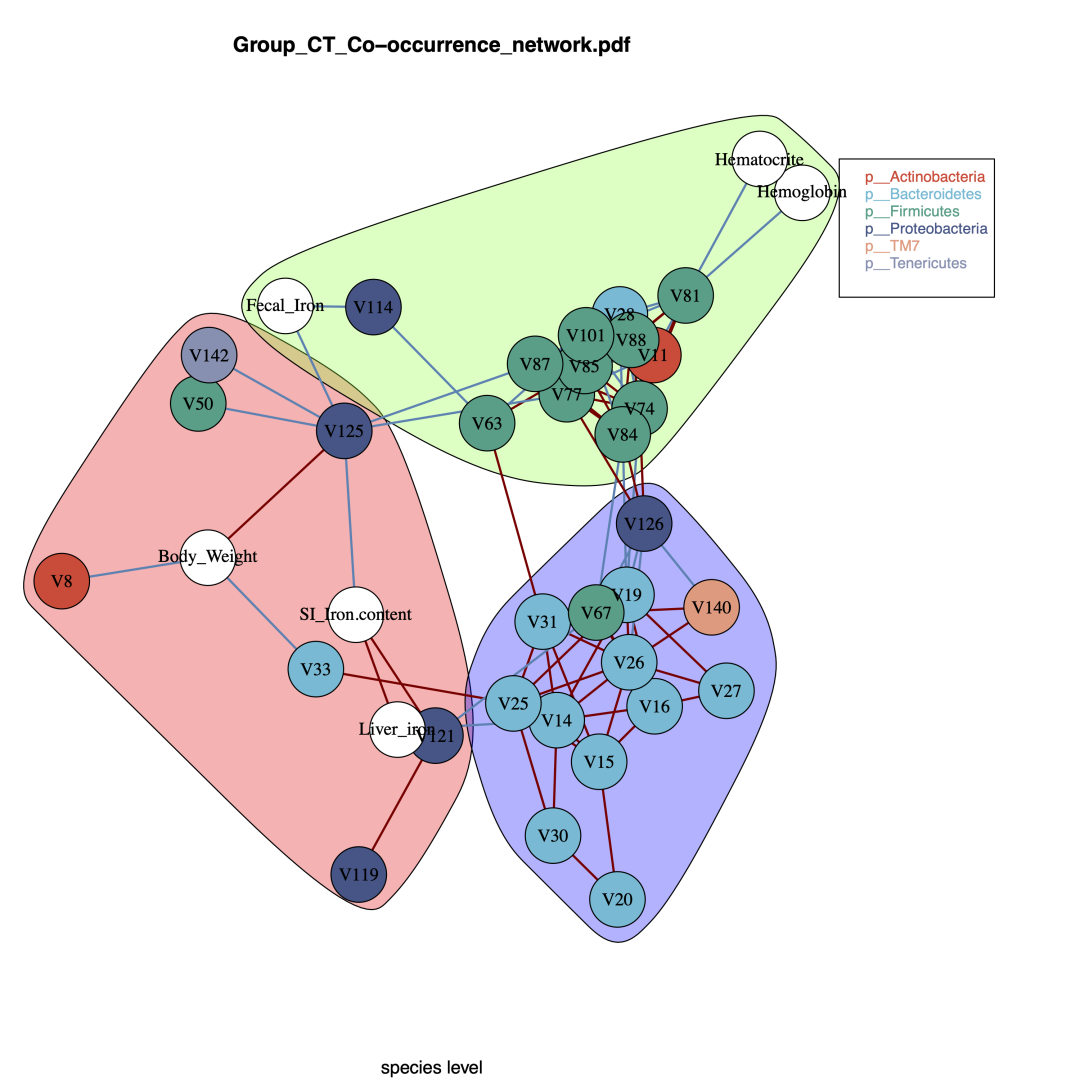
cooc5.png
cooc6.png
### 5.5 Structure结构图
物种结构图常用来展现样本主要微生物的分布情况,多用堆叠柱状图的形式出现。本章节`structure_plot`模块可以基于平均值、中位数、最大丰度、最小丰度的四种方式筛选TOP物种,并进行结构图展示。
```R
library(EasyMicroPlot) # 加载包
# 基本使用方式,自动按照均值前10名绘制物种结构图
structure_re <- structure_plot(dir = '16s_data/',design = 'mapping/mapping.txt',
min_relative = 0.001,min_ratio = 0.7,num=10)
##structure_re$result$filter_data 这里存储了前置data_filter函数过滤的核心微生物结果
##structure_re$result$top_abundance 这里存储了结构图中TOP显示物种的丰度数据
## structure_re$pic 这里存储了各个微生物物种级别及各组的结构图
structure_re$pic$class$barplot$Total # 以纲为级别做为示例
```
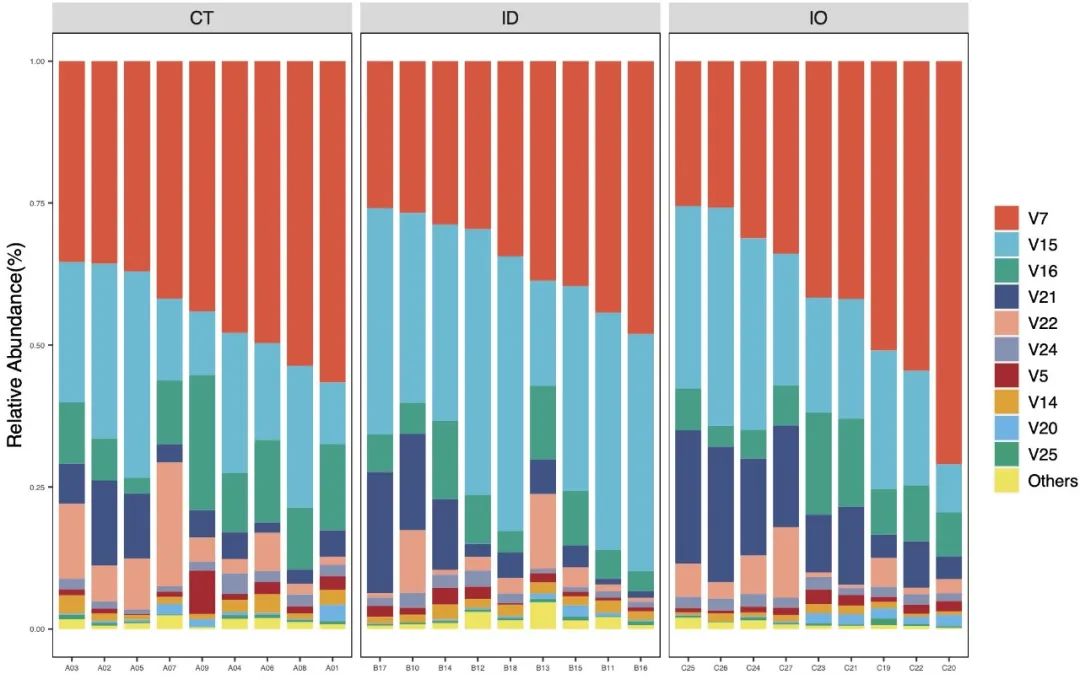
str_bar1.png
```R
# 图形输出可以通过多个参数对图形进行美化
top_num <- 5 ## 选取前五名物种
group_order <- c('ID','CT','IO') ## 设定组排序,名称需要绝对一致
tax_order <- c('Others','V7','V15','V16','V21','V22') ## 设定物种排序,名称需要绝对一致
## 自定义颜色方案,颜色数目需与物种数目一致
cols <- c("#432c39", "#437478", "#b8b7a1", "#eed7b8","#e5855f", "#db6c4e", "#d24d3a")
# mytheme 支持ggplot2主题语法
library(ggplot2)
newtheme_slope=theme(axis.text.x =element_text(angle = 45, hjust = 1,size = 10))
# 出图
# 注意由于设置了只符合纲级别的tax_level参数,因此建议只输入纲级别数据,否则其他级别数据将会因无法匹配而出现warning
# 这里可以利用pattern参数,根据文件名只选择读取纲级别数据;也可以利用预读取纲级别数据,再利用data参数输入
# measure参数 可以选择图形中某个物种按照丰度由低到高进行样本排序
structure_re <- structure_plot(dir = '16s_data/',design
= 'mapping/mapping.txt',
min_relative = 0.001,min_ratio = 0.7,num=top_num,
group_level = group_order,tax_level =tax_order,palette = cols,
mytheme =newtheme_slope,pattern = 'L3',measure = 'Others')
structure_re$pic$class$barplot$Total
```
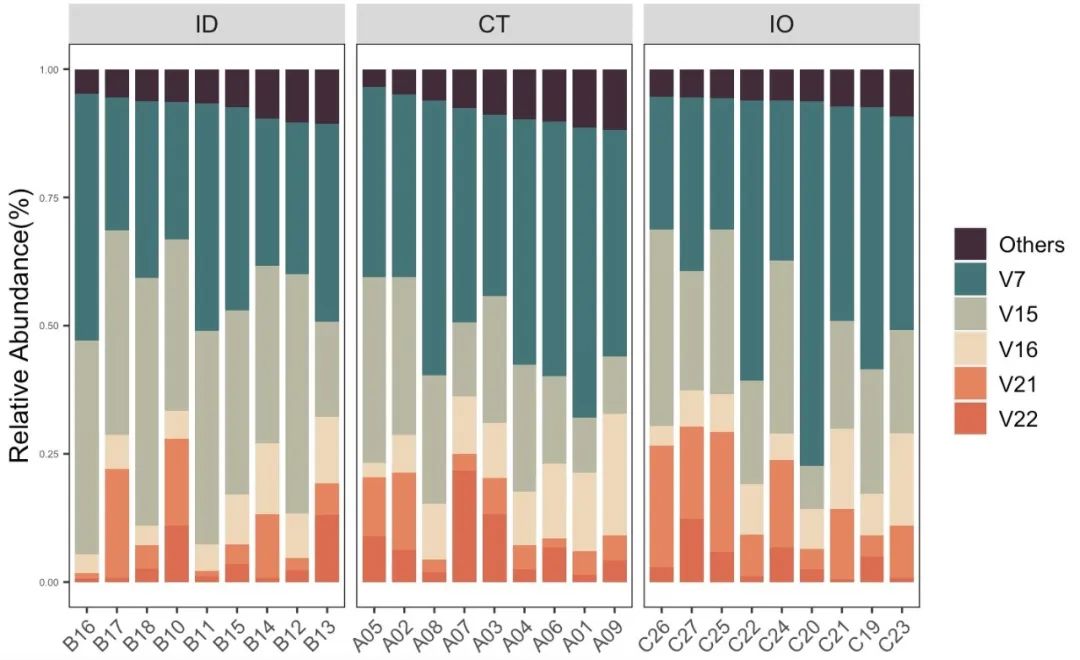
str_bar2.png
### 5.6 微生物差异箱型图
微生物物种差异箱型图
```R
# 首先利用过滤核心模块读取原始数据
library(EasyMicroPlot) # 加载包
core_data <- data_filter(dir = '16s_data/',design
= 'mapping/mapping.txt',
min_relative = 0.001,min_ratio = 0.7)
# 选择出自己需要的微生物级别数据,此处以种级别为例
sp <- core_data$filter_data$species
# 利用函数进行计算,这里选择V14,V15,V15用于示例
# method 也可以选择ttest进行两两T检验
tax_re <- tax_plot(data = sp,tax_select
= c('V14','V15','V16'),
method = 'LSD',width = 5,height = 5)
## tax_re$pic 这里存储了基本图形结果
## tax_re$html 这里存储交互式图形结果
## tax_re$Post_Hoc # 当method选择onewayANOVA的多重检验时,这里存储了事后检验的详细结果
tax_re$pic$total
```
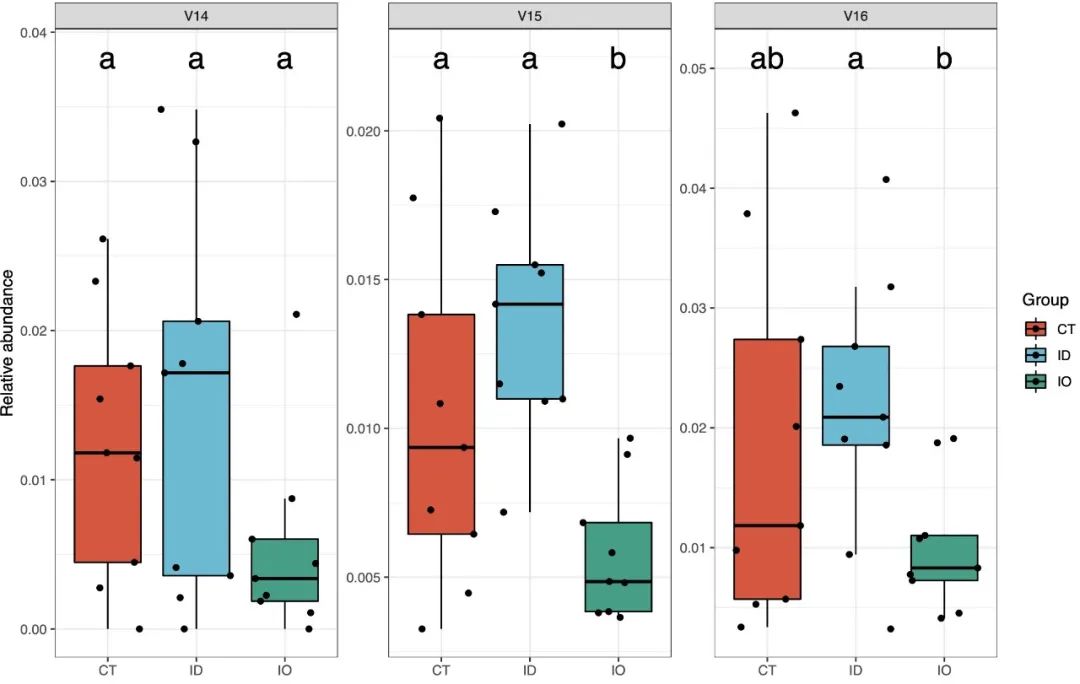
taxplot1.png
### 5.7 随机森林交叉验证模型
近年来主流的微生物分析中,随机森林模型常被用来进行关键微生物的筛选。但是由于随机数导致模型结果的不确定性以及难以确定最优价值的差异菌种,因此随机森林交叉验证递归剔除弱重要性的策略可以帮助研究者在一定程度下筛选出一批具有潜在重要性的关键菌种。本章节`RFCV`模块提供了一种快速根据核心微生物进行各个级别随机森林模型快速筛选的方法及可视化结果。
```R
# 首先利用过滤核心模块读取原始数据
library(EasyMicroPlot) # 加载包
core_data <- data_filter(dir = '16s_data/',design
= 'mapping/mapping.txt',
min_relative = 0.001,min_ratio = 0.7)
# 选择出自己需要的微生物级别数据
RF_data <- core_data$filter_data$species
## 进行随机森林交叉验证模型计算
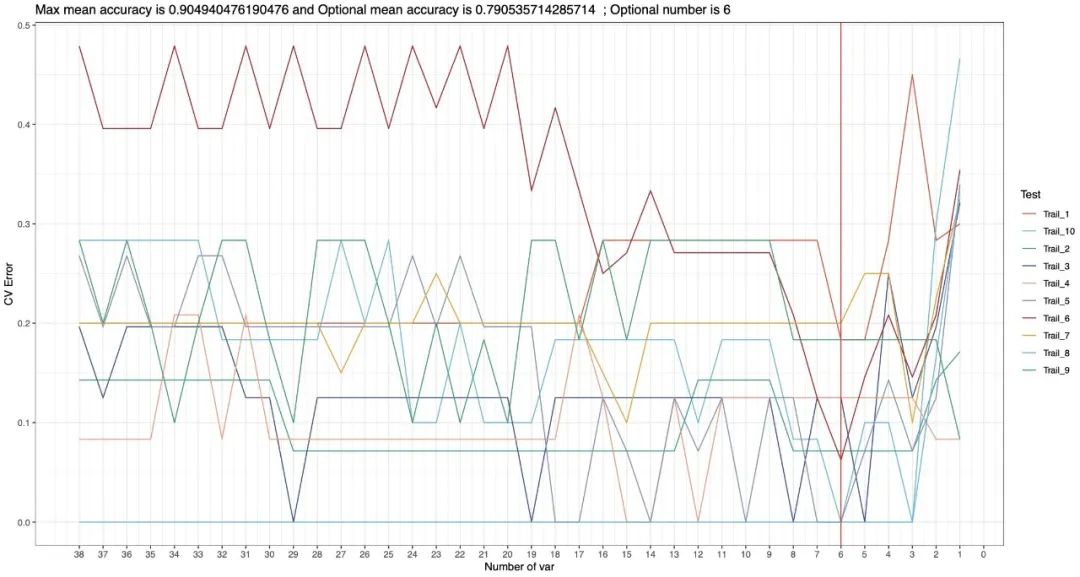
# 本模块根据交叉验证中不同随机数种子下平均错误率和不同随机数下错误率标准差判定最适宜截线
RF_re <- RFCV(RF_data)
## RF_re$RFCV_data 输入的微生物数据
## RF_re$RFCV_result #随机森林交叉验证的基本结果
RF_re$RFCV_result_plot$intersect_num #不同随机数下最适宜结果的交集物种
RF_re$RFCV_result_plot$union_num #不同随机数下最适宜结果的并集物种
RF_re$RFCV_result_plot$curve_plot ## RFCV平均错误率下降图
```
RFCV1.png
由于随机森林判断的最优特征不一定具有统计学意义,因此可以利用```tax_plot```函数快速查看各组间差异结果。这里建议采用不同随机数下并集的结果进行统计。
```R
# 快速计算潜在关键菌的统计学差异
# 这里也可以 method= ttest 采用T检验的方法判断
tax_re <- tax_plot(RF_data,tax_select = RF_re$RFCV_result_plot$union_num,method = 'LSD')
tax_re$pic$total
```
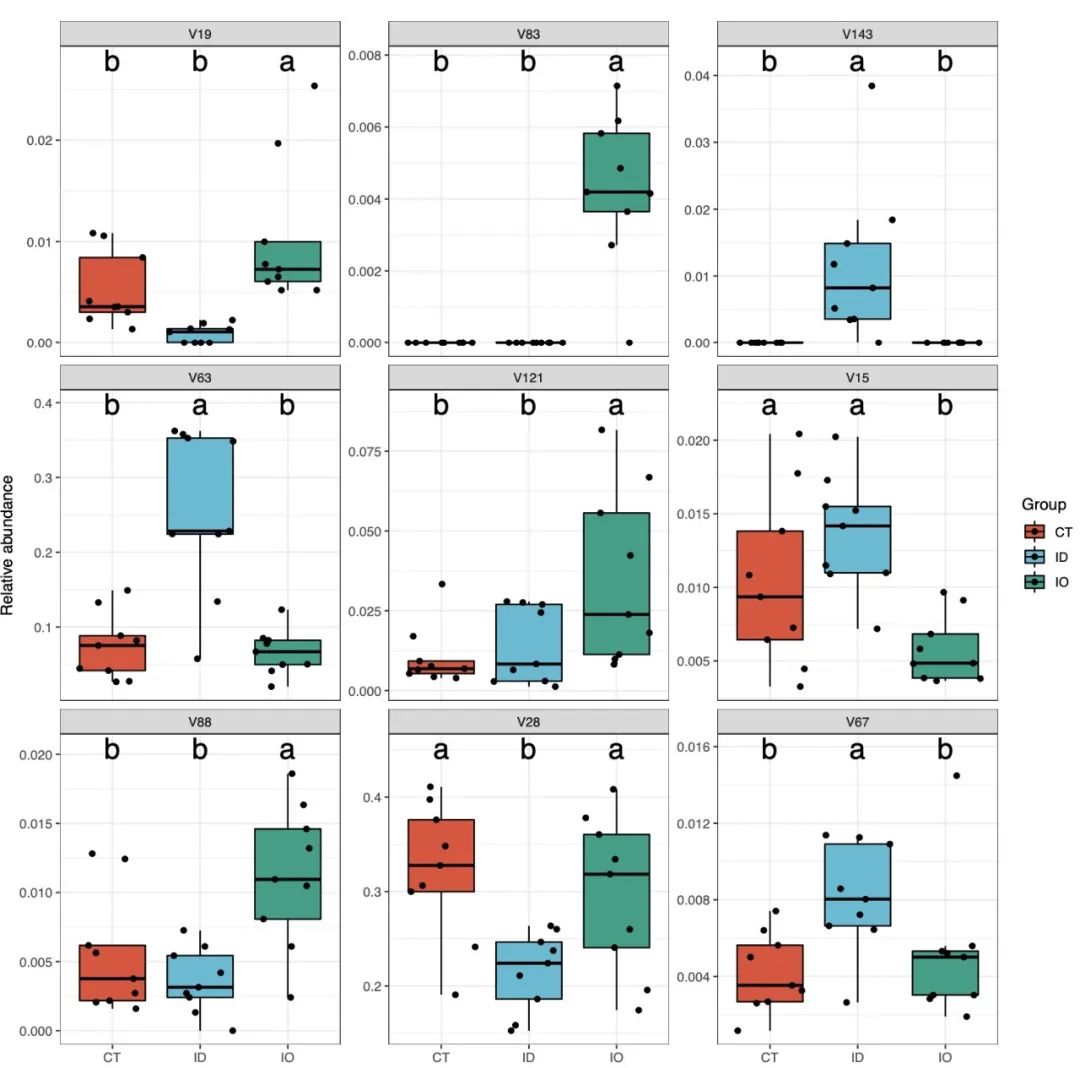
RFCV3.png
如果我们的输入数据为二分类数据,例如高血压组和非高血压组,糖尿病组和非糖尿病组等,可以根据模型结果进行ROC评估AUC面积。
```R
# 由于本示例数据为三组,分别为CT(正常饲料组),IO(铁剂过量饲料组)和ID(铁剂缺乏饲料组),无法直接使用与二分类的ROC模型评估
# 因此可以将数据转为二分类数据,例如CT组和非CT组
RF_data_binary
<- RFCV_data_binary(RF_data,rf_estimate_group
= 'CT',id_not = 'NOT_CT')
# 再次进行RFCV计算
RF_re2 <- RFCV(RF_data_binary)
# 将RFCV的结果直接带入函数
# rf_tax_select 为选取所需要进一步评估的物种,这里可以直接带入上并集结果
RFCV_roc(RF_re,rf_tax_select
= F_re2$RFCV_result_plot$union_num,rf_estimate_group = 'NOT_CT')
```
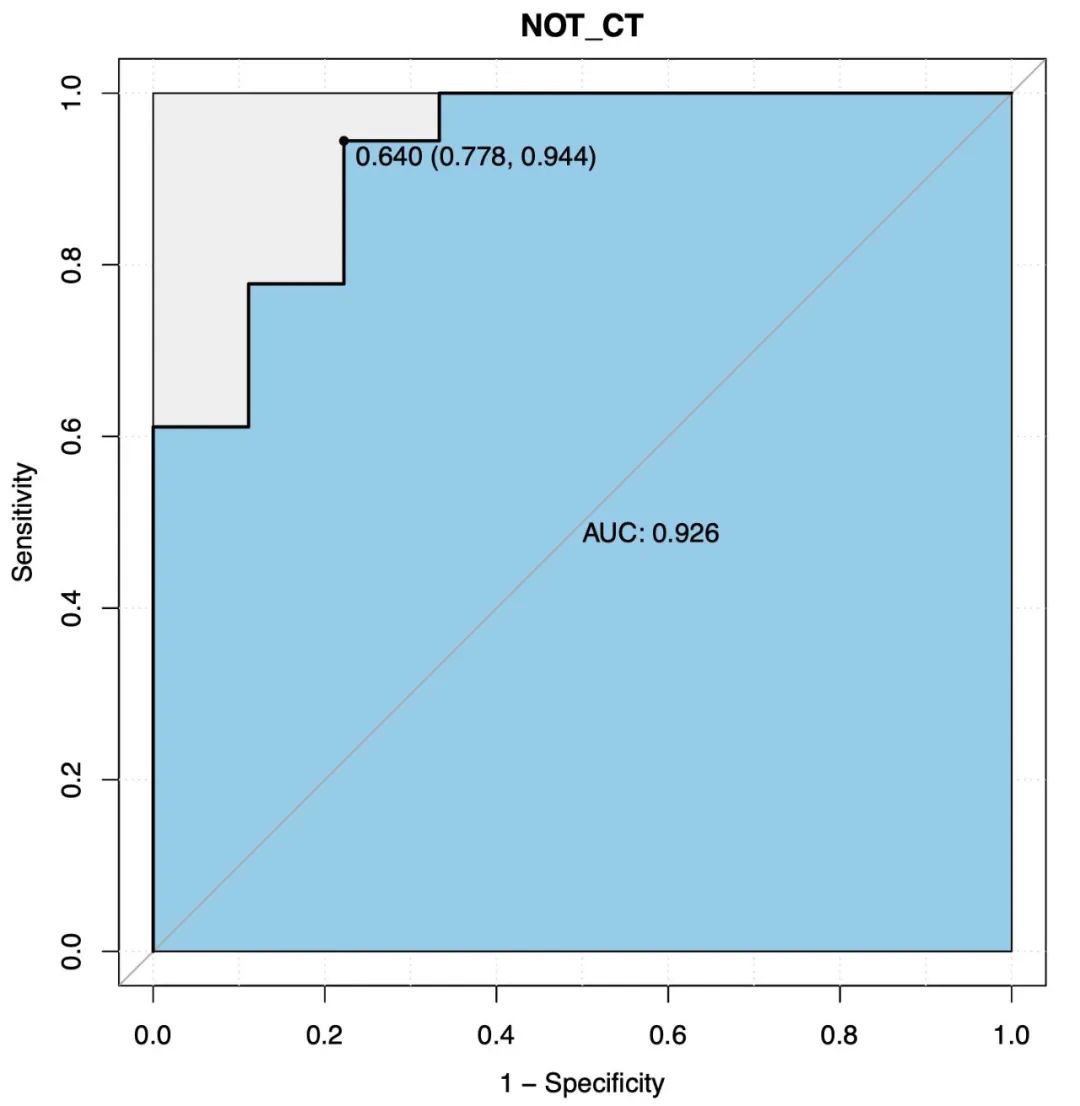
RFCV4.png
```R
# 也可以根据tax_plot函数的结果,手动挑选合适的物种纳入ROC评估
# 例如可以挑选出具有统计学差异的核心微生物物种
tax_re <- tax_plot(RF_data_binary,tax_select
= RF_re2$RFCV_result_plot$union_num,method = 'ttest')
tax_re$pic$total
RFCV_roc(RF_re2,rf_tax_select
= c('V83','V121','V143'),rf_estimate_group = 'NOT_CT')
```
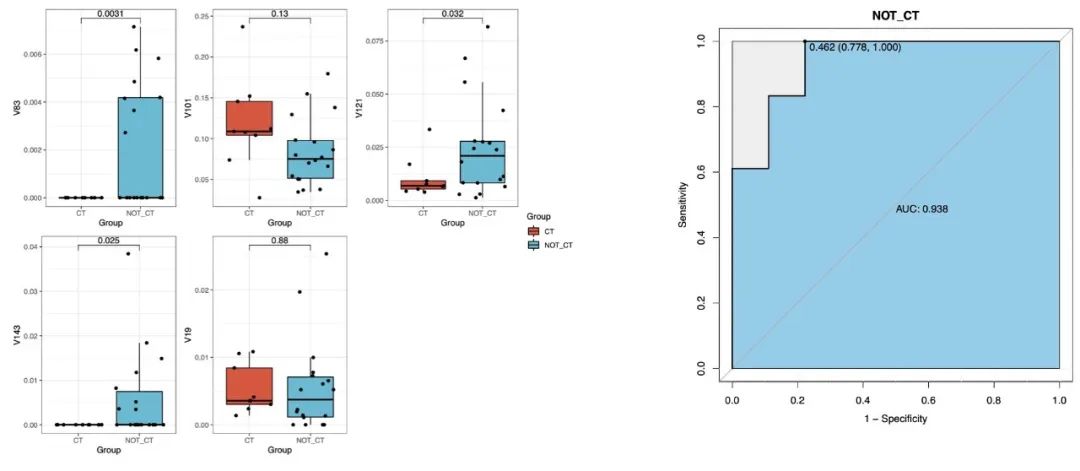
RFCV7.png
### 5.8 相关性分析图
微生物研究中,微生物数据常常和表型数据存在着广泛的线性关系。本章节`EMP_COR`模块提供两种展现微生物数据和表型数据相关性的方式。
**注意**:当表型数据和微生物数据样本不完全一致时,用户无需手动修改原始数据,此模块将自动选择二者交集进行计算。
```R
# 基本代码
library(EasyMicroPlot) # 加载包
# 加载表型数据
data(EMP)
meta_data <- EMP$iron # 这里使用内置的表型数据,用户也可以自行读取自己的数据,注意满足3.2格式要求
core_data <- data_filter(dir = '16s_data/',design
= 'mapping/mapping.txt',
min_relative = 0.001,min_ratio = 0.7)
core_phylum <- core_data$filter_data$phylum # 这里示例为门级别,用户可以自行选择所需要的微生物物种级别
# 两种不同相关性计算方案
# method 可以选择pearson,kendall和spearman相关性检验方法
## 微生物数据和表型数据分开进行关联分析 (热图形式)
## cor_output = T 可以将图形自动输出到本地工作区
cor_re <- EMP_COR(data
= core_phylum,meta=meta_data,cor_output = F,
method = 'spearman',aes_value = 1)
## 微生物数据和表型数据合并进行关联分析 (三角图形式)
cor_re <- EMP_COR(data
= core_phylum,meta=meta_data,cor_output = F,
method = 'spearman',aes_value = 2)
cor_re$r ## 相关性系数结果
cor_re$p ## 相关性检验结果
```
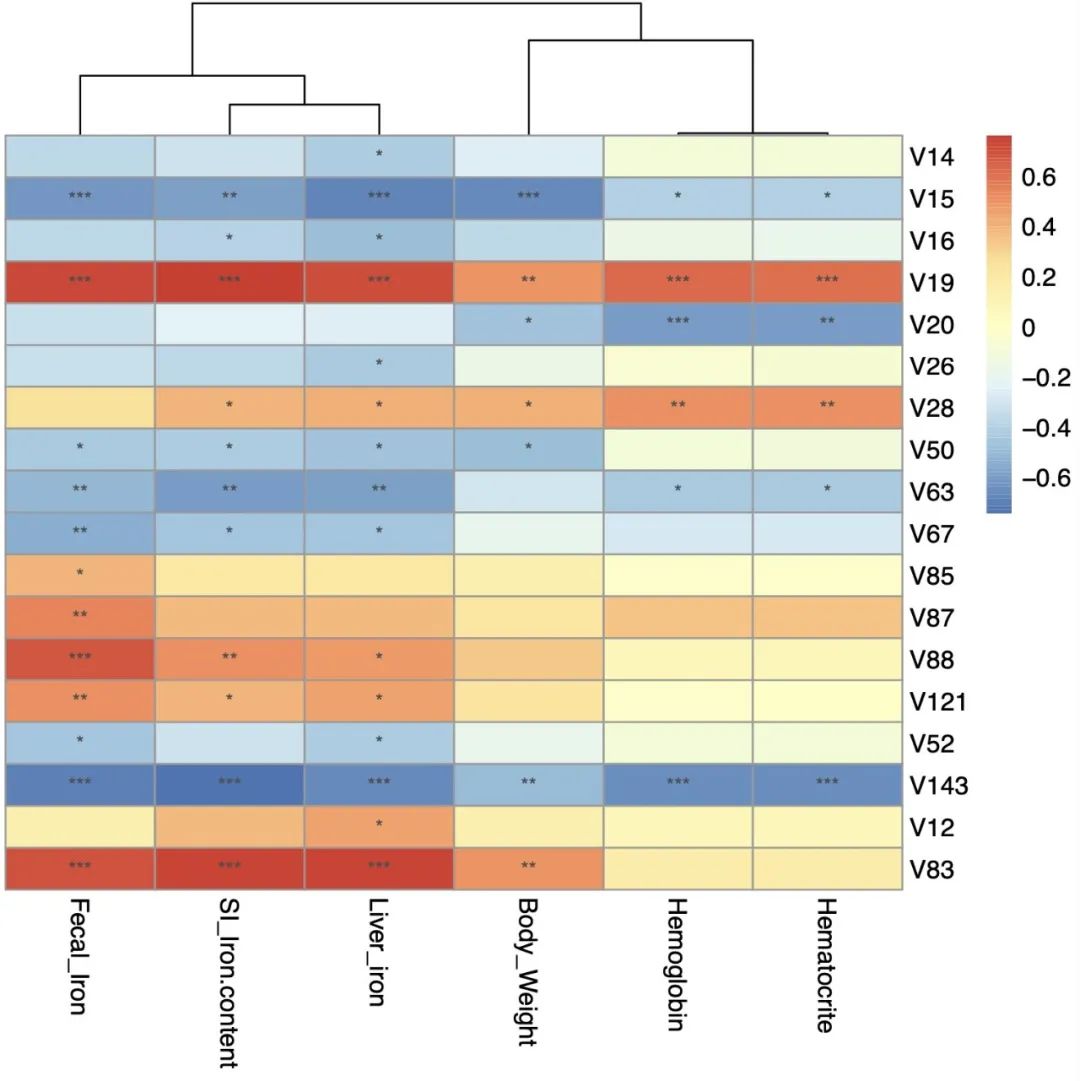
EMP_COR1.png
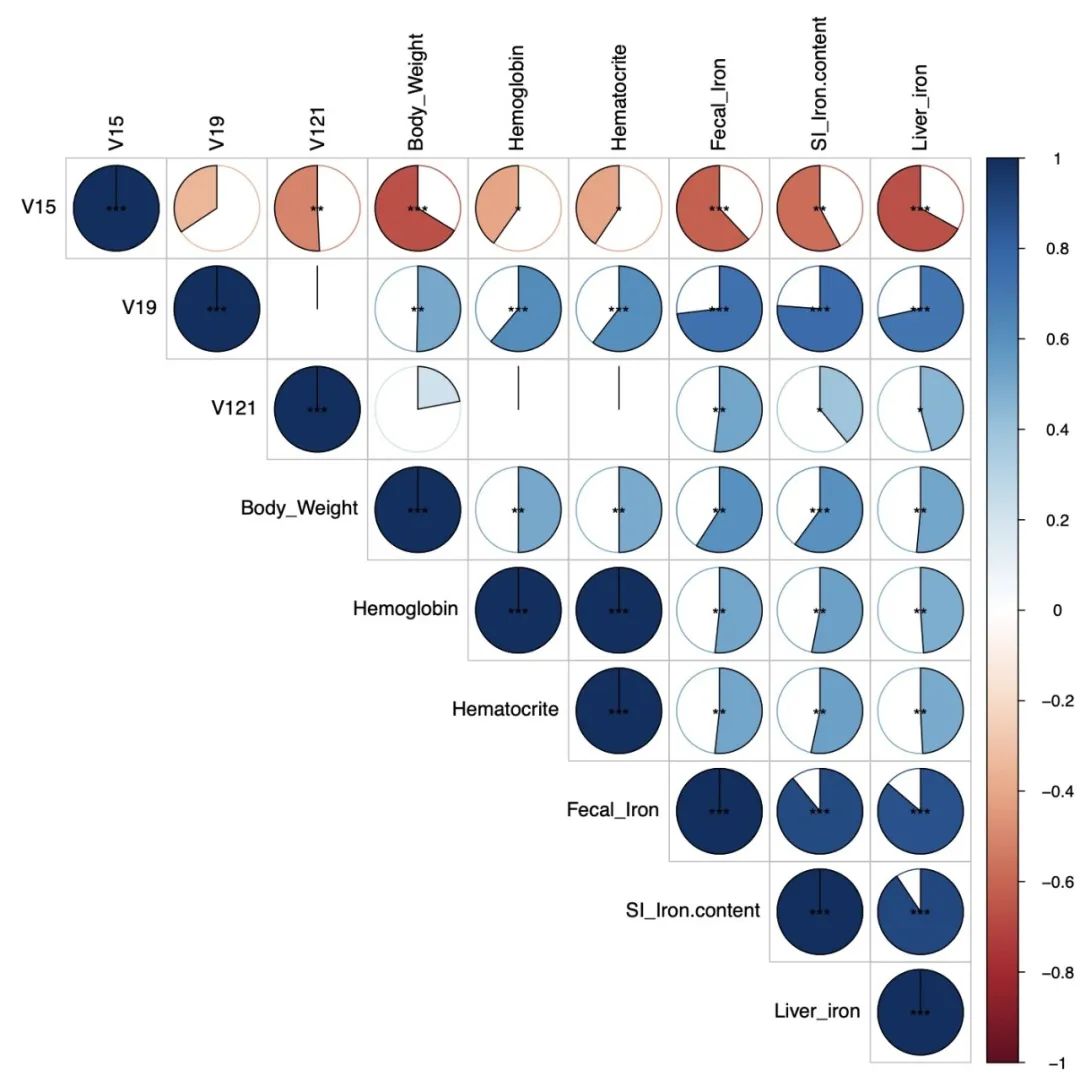
EMP_COR2.png
### 5.9 双变量拟合图
本章节提供`EMP_COR_FIT`模块提供可双变量线性拟合的模型及可视化结果。
```R
# 基本代码
library(EasyMicroPlot) # 加载包
# 加载表型数据
data(EMP)
meta_data <- EMP$iron # 这里使用内置的表型数据,用户也可以自行读取自己的数据,注意满足3.2格式要求
core_data <- data_filter(dir = '16s_data/',design = 'mapping/mapping.txt',
min_relative = 0.001,min_ratio = 0.7)
core_species <- core_data$filter_data$species # 这里用户可以自行选择所需要的微生物物种级别
# 一元一次拟合方程
# se = T 可以增加拟合置信区间
fit_result <- EMP_COR_FIT(data=core_species,meta = meta_data,var_select = c('V19','SI_Iron.content'),
formula = y~poly(x,1,raw = T),width = 5,height = 5,se = F,group = F,eq_size = 3)
fit_result$pic # 基本拟合图形结果
fit_result$html # 交互式拟合图形结果
```
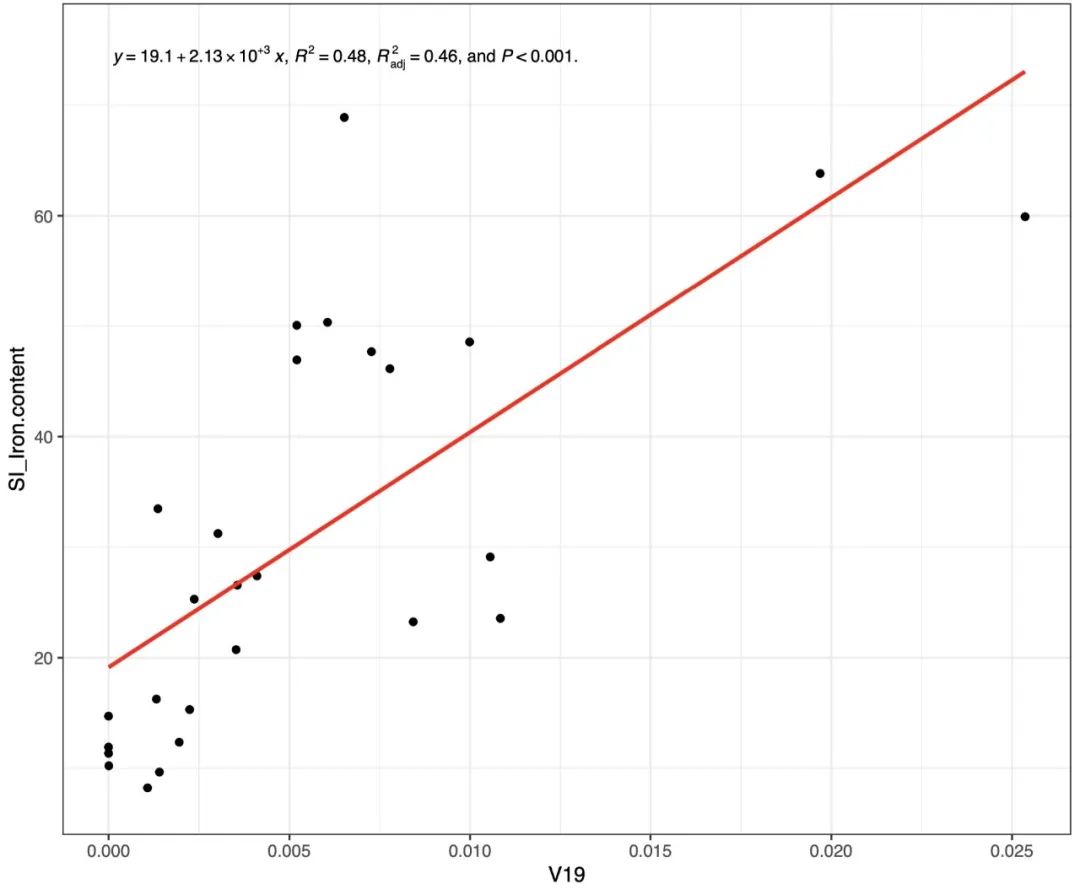
EMP_COR_FIT1.png
```R
# 一元二次拟合方程
fit_result <- EMP_COR_FIT(data=core_species,meta = meta_data,var_select = c('V19','SI_Iron.content'),
formula = y~poly(x,2,raw = T),width = 5,height = 5,se = F,group = F,eq_size = 3)
fit_result$pic
fit_result$html # 交互式html
```
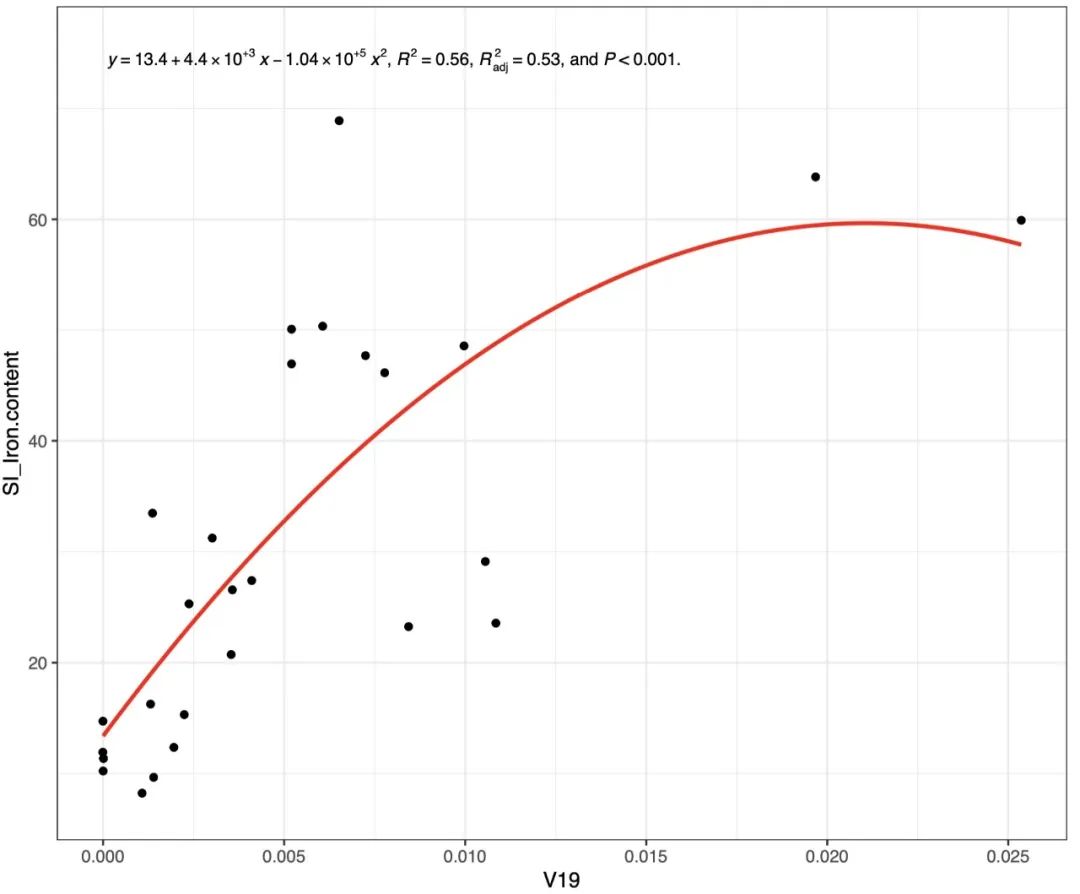
EMP_COR_FIT2.png
```R
# 也可以用group = T 将数据按照分组进行拟合
# 当方程式过长导致显示不全时,可以用eq_size调整方程式的字体大小
fit_result <- EMP_COR_FIT(data=core_species,meta = meta_data,var_select = c('V19','SI_Iron.content'),
formula = y~poly(x,1,raw = T),width = 5,height = 5,se = T,group = T,eq_size = 3)
fit_result$pic
fit_result$html # 交互式html
```
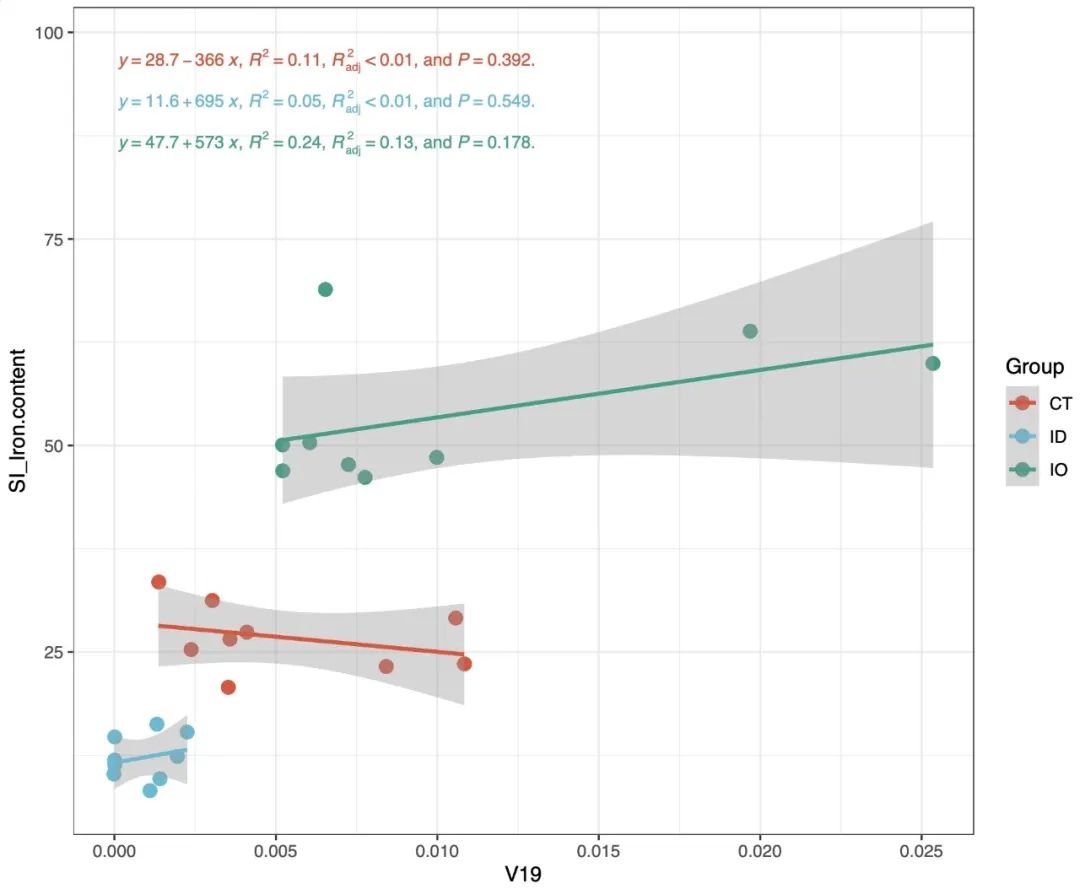
EMP_COR_FIT4.png
### 5.10 冗余分析
在微生物研究中,RDA分析常被用来评估微生物数据和表型数据之间的相互关系。
```R
# 基本代码
library(EasyMicroPlot) # 加载包
# 加载表型数据
data(EMP)
meta_data <- EMP$iron # 这里使用内置的表型数据,用户也可以自行读取自己的数据,注意满足3.2格式要求
core_data <- data_filter(dir = '16s_data/',design = 'mapping/mapping.txt',
min_relative = 0.001,min_ratio = 0.7)
core_species<- core_data$filter_data$species # 这里用户可以自行选择所需要的微生物物种级别
RDA_re <- EMP_COR_RDA(data = core_species,meta=meta_data,width = 8,height = 8)
RDA_re$input_data # RDA模型输入的数据
RDA_re$model #RDA模型的基本结果
# RDA模型的置换检验结果
RDA_re$model_information$model_permutest
# 表型数据因子检验结果
RDA_re$model_information$model_envfit
# 表型数据膨胀系数检验
RDA_re$model_information$model_vif
# 基本图形展示
RDA_re$plot$pic
RDA_re$plot$html # 交互式模式
```
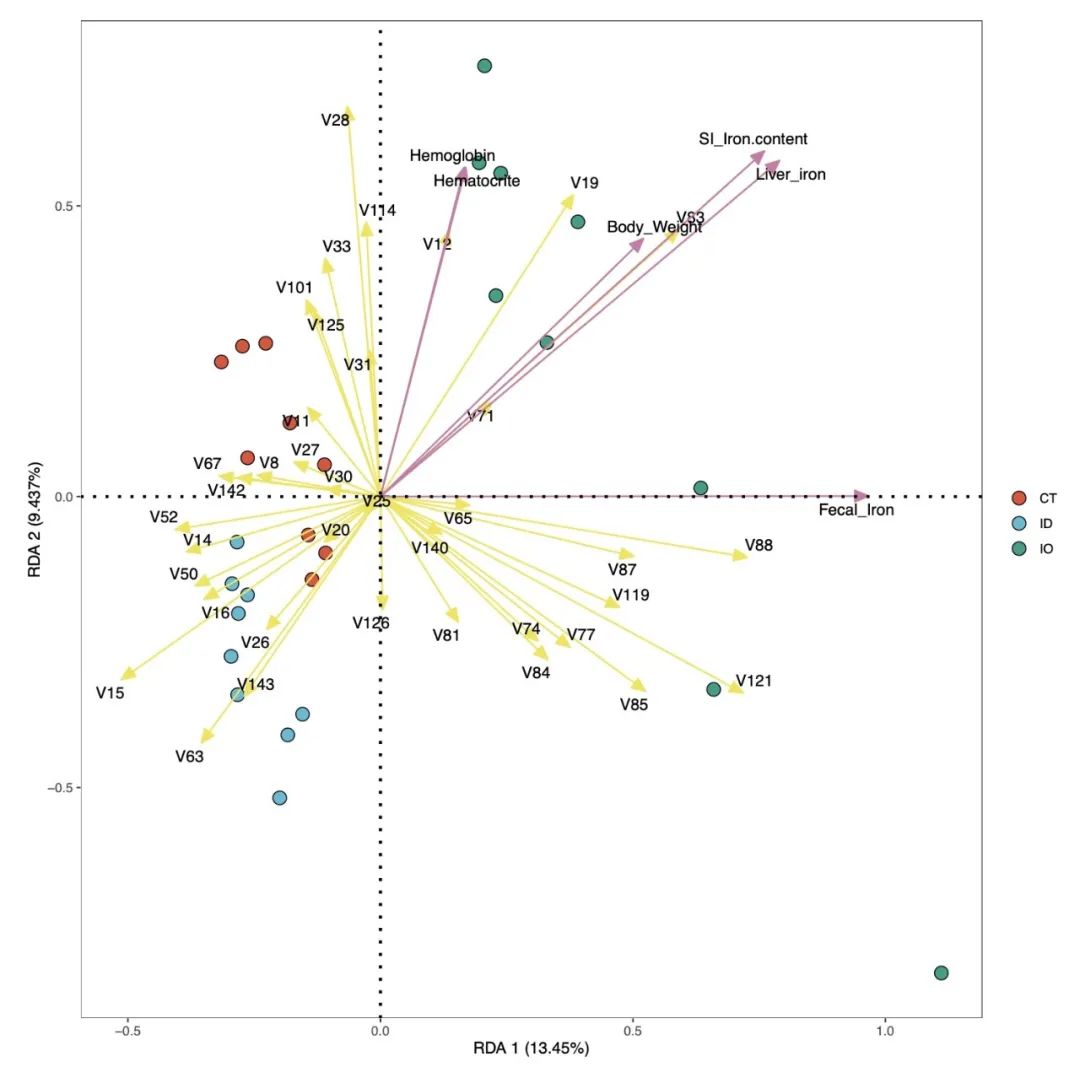
RDA1.png
```R
# 可以调整参数,进一步美化或者调整图形输出效果
# zoom 参数调整箭头和样本的相对距离
# arrow_col 调整两个箭头的颜色
# palette 调整分组颜色
# ellipse 在0~1之间取值标注样本置信区间
RDA_re <- EMP_COR_RDA(data = core_species,meta=EMP$iron,
width = 10,height = 10,ellipse = 0.7,zoom = c(1,1.5,2),
arrow_col = c('#65776b','#e26354'),palette = c('#a14a8a','#fdce72','#459bce'))
RDA_re$plot$pic
```
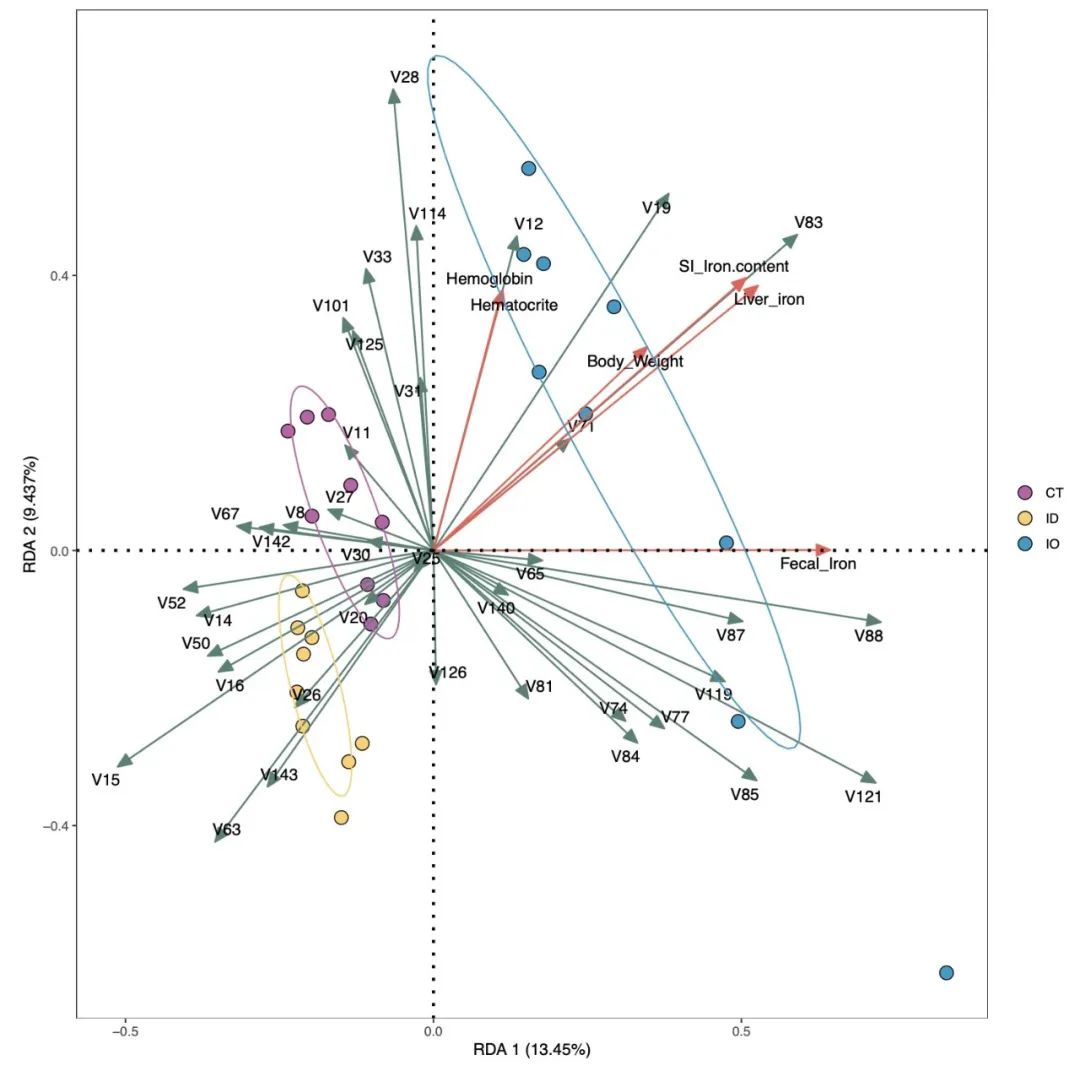
RDA2.png
### 5.11 多重相关性Sankey图
Sankey图过去常被用来描述归属的流动关系,近年来有研究利用sankey图来表现多层数据之间的相互关系。例如第一层为微生物数据,第二层为微生物功能数据(picrust结果或者功能基因组装结果),第三层为样本表型数据(临床症状等)。这种分层相互关系的展现形式,可以很好的描述微生物丰度-微生物功能-宿主病理生理表型的逻辑关系。本章节`EMP_COR_SANKEY`模块可以基于数据框列表,快速绘制多重相关性的交互式Sankey图。
注意 1:`data_list`支持多层数据,只需按照顺序依次排列在`list`内。
注意 2:`data_list`内数据框包含的样本不必完全一致,每两层之间的相互关系将采用交集样本计算。
注意 3:`EMP_COR_SANKEY`模块计算相互关系时,将根据`rvalue`和`pvalue`过滤掉不符合意义的边,而孤立的节点将会被去除。
注意 4:`EMP_COR_SANKEY`模块计算相互关系时,将判定中间层的节点必须左右均具有符合条件的相互关系,如只存在一侧关系的中间层节点将会被自动过滤。
```R
# 基本用法
library(EasyMicroPlot) # 加载包
data(EMP) # 加载内置示例数据
Sankey_pic<- EMP_COR_SANKEY(data_list = EMP$Sankey_data,rvalue = 0.3,pvalue = 0.05)
Sankey_pic$sankey_data # 分层相关性结果
Sankey_pic$plot # 交互式图形结果
```
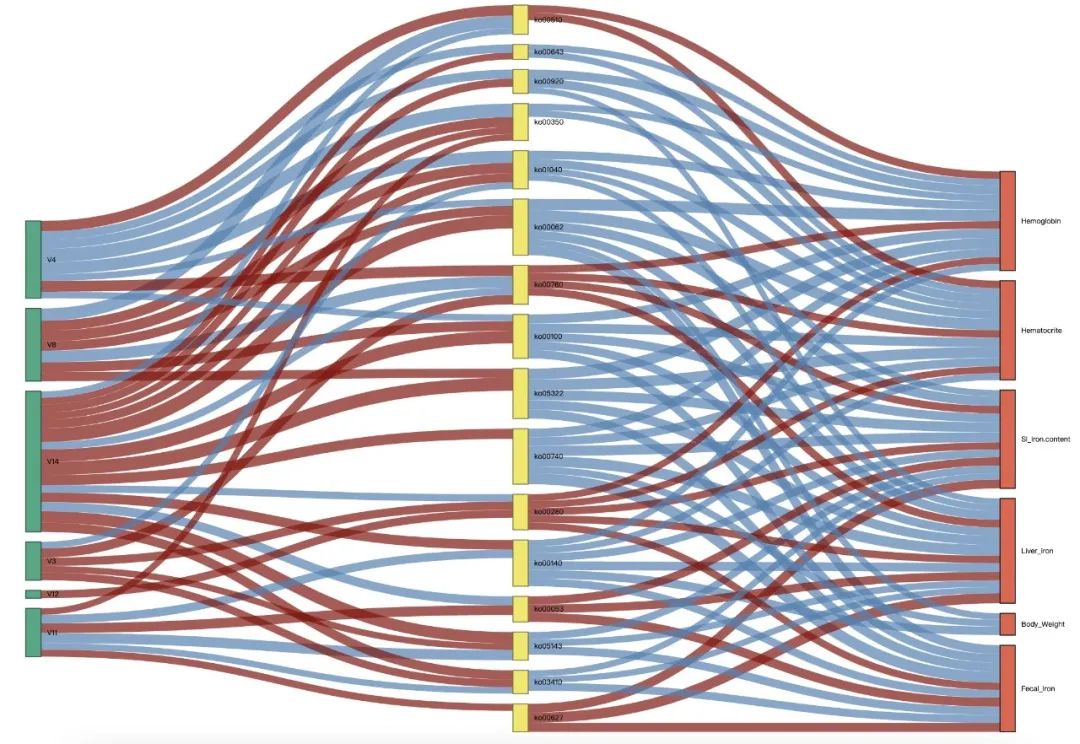
sankey.png
### 6 总结
近年来随着微生物领域的火热发展,越来越多的生物信息学工具涌现而出,特别是各种R包和脚本有效的帮助了相关领域研究人员处理微生物信息数据。EasyMicroPlot包由广东省科学院微生物研究所谢黎炜教授团队(xielw@gdim.cn)开发,致力于辅助低代码经验研究人员快速便捷高效的进行微生物下游数据分析。目前EMP包已经发表,更多的功能模块仍在进一步优化和整合中。
猜你喜欢
iMeta简介 高引文章 高颜值绘图imageGP 网络分析iNAP
iMeta网页工具 代谢组MetOrigin 美吉云乳酸化预测DeepKla
iMeta综述 肠菌菌群 植物菌群 口腔菌群 蛋白质结构预测
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature
一文读懂:宏基因组 寄生虫益处 进化树 必备技能:提问 搜索 Endnote
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流快速解决科研困难,我们建立了“宏基因组”讨论群,己有国内外6000+ 科研人员加入。请添加主编微信meta-genomics带你入群,务必备注“姓名-单位-研究方向-职称/年级”。高级职称请注明身份,另有海内外微生物PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。
点击阅读原文,跳转最新文章目录阅读


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









