






 本文介绍基于培养组的宏基因组学(CBM)策略,用于挖掘沙漠土壤微生物暗物质。CBM整合大规模分离培养与宏基因组测序,捕获直接测序遗漏的分类和功能多样性,揭示大量候选细菌新分类单元。但CBM不能反映微生物真实组成与丰度,结合直接测序可深入了解沙漠微生物组。
本文介绍基于培养组的宏基因组学(CBM)策略,用于挖掘沙漠土壤微生物暗物质。CBM整合大规模分离培养与宏基因组测序,捕获直接测序遗漏的分类和功能多样性,揭示大量候选细菌新分类单元。但CBM不能反映微生物真实组成与丰度,结合直接测序可深入了解沙漠微生物组。
利用基于培养组的宏基因组学和高分辨分析捕获沙漠土壤微生物暗物质
Capturing the microbial dark matter in desert soils using culturomics-based metagenomics and high-resolution analysis

Article,2023-9-23,npj Biofilms and Microbiomes, [IF 9.2]
DOI:https://doi.org/10.1038/s41522-023-00439-8
原文链接:https://www.nature.com/articles/s41522-023-00439-8#citeas
第一作者:李帅 (Shuai Li)
通讯作者:李文均 (Wen-Jun Li);董雷 (Lei Dong)
合作作者:连文慧;韩嘉瑞;Mukhtiar Ali;林志亮;刘永红;李丽;张东亚;蒋先芝
主要单位:
中山大学生命科学学院
嘉应学院生命科学学院
中国科学院新疆生态与地理研究所
慕恩生物微生物组研究中心
- 摘要 -
荒漠(Deserts)占地球陆地表面积的1/3,是微生物多样性的潜在重要宝库,然而其中的绝大多数微生物仍未被发现和表征,被视为“微生物暗物质”。在此,我们介绍了一种多组学策略——基于培养组的宏基因组学(Culturomics-based Metagenomics,CBM),其整合了大规模分离培养,以及基于全长16S rRNA 基因和鸟枪法的宏基因组测序。结果表明,通过提高ASVs和高/中质量MAGs的恢复率,CBM捕获了直接测序所遗漏的巨大分类和功能多样性。值得注意的是,CBM允许事后恢复感兴趣的微生物(如新分类单元或一些特定类群),甚至是那些在培养物中丰度极低的类群。此外,基于CBM和直接测序的菌株水平分析结果表明,沙漠土壤中蕴藏着大量候选细菌新分类单元(1941个,占比51.4%),其中1095个(来自CBM)是可培养的。然而,CBM并不能准确地反映微生物在原位环境中的真实组成与功能相对丰度,将CBM与直接的宏基因组测序相结合,可以更深入地了解沙漠微生物组。总之,这项研究揭示了高分辨的CBM策略是深入探索沙漠土壤中未开发细菌新资源的理想方法,极大地扩展了我们对隐匿于广袤沙漠中的微生物暗物质的认识。
【注:沙漠是荒漠生境中最具代表性的类型之一,通常是指“沙质荒漠”,沙漠地表多覆盖有沙丘或沙滩。沙漠在全球分布范围十分广泛且覆盖面积巨大,地球表面的陆地面积约为1.62亿平方千米,其中大约1/3的地区为荒漠,1/5的地区属于沙漠。】
- 引言 -
荒漠约占全球陆地总面积的1/3,是地球上面积最大、研究最不充分的生境之一,同时也是地球微生物多样性的重要宝库。荒漠微生物在维持生态稳定和生物地球化学循环方面发挥着重要作用。对荒漠生态系统的微生物多样性、组成和功能进行剖析,将有助于了解旱地生命带来的全球变化、威胁与机遇。许多研究表明,作为荒漠区的一种典型代表类型,沙漠生境中许多不同的微生物可以产生一系列新的生物活性化合物,包括抗菌、消炎、抗肿瘤及群体感应抑制等诸多方面。然而,由于生态位特化性强、样品采集困难以及传统培养方法的适应性有限等因素,绝大多数沙漠微生物被忽视了在实验室条件下的培养与分类鉴定。这一大部分尚未被充分探索的微生物多样性,代表了巨大的未开发、未表征的微生物资源,俗称“微生物暗物质(Microbial Dark Matter)”,其从根本上阻碍了微生物生态学的发展和生物资源的开发利用。
宏基因组学技术(如16S rRNA扩增子、鸟枪法测序)提供了相对简单快速的方法,用于分析环境样本中微生物群落的分类学组成和功能潜力,恢复微生物的全基因组序列,而无需进行分离培养实验。近年来,对沙漠微生物组进行的宏基因组学研究极大推动了我们目前对全球沙漠(如阿塔卡马沙漠、纳米布沙漠、内盖夫沙漠、古尔班通古特沙漠和极地沙漠等)微生物物种组成和功能的了解,为进一步深入探索沙漠微生物资源奠定了重要基础。然而,环境样本宏基因组数据的实用性与多种因素密切相关,例如微生物群落的复杂度和生物量、测序技术和参考数据库等,这些均可能会导致某些特定类群(如低丰度类群)在直接测序中被忽略。
培养组学,是一种利用多重分离培养条件结合16S rRNA基因测序和/或其它技术(如MALDI-TOF)的微生物组研究方法,其应用大大提高了我们对可培养微生物多样性的认知。尽管培养组学在获得微生物纯培养物方面有很大作用,但它通常被认为是劳动和资源密集型的,且可能会遗漏群落中的一些重要特定类群。因此,通过系统的培养组学获得全面的菌株数据集仍是一项重要而尚未解决的挑战。
值得注意的是,选择性培养富集以降低群落复杂性的策略可能有助于特定环境中微生物群落(如肠道)的宏基因组学研究。然而,还未有结合使用培养组学(Culturomics)和培养富集宏基因组测序(Culture-enriched metagenomic sequencing)两种方法来研究沙漠微生物组的报道。鉴于现有知识,我们假设基于大规模培养组学方法和高分辨分析的富集培养宏基因组测序能在很大程度上填补直接测序中土壤微生物组的缺失部分。考虑到这一点,我们的主要目的是评估结合培养组学和宏基因组学的多组学策略在沙漠土壤微生物组研究中的有效性和前景,并为探索沙漠土壤中的微生物暗物质提供新的视角。
以挖掘沙漠土壤微生物暗物质为例,我们提出了一种融合了培养组学和宏基因组学方法(全长16S扩增子和鸟枪法测序)的综合策略,即基于培养组的宏基因组学(Culturomics-based metagenomics,CBM)(图1)。我们的研究结果揭示了沙漠土壤中细菌新资源潜力的未被描述的景观,并展示了CBM在提高沙漠微生物组的分类和功能分辨率方面的巨大优势,这将扩大对隐匿在沙漠中的微生物暗物质的认识。
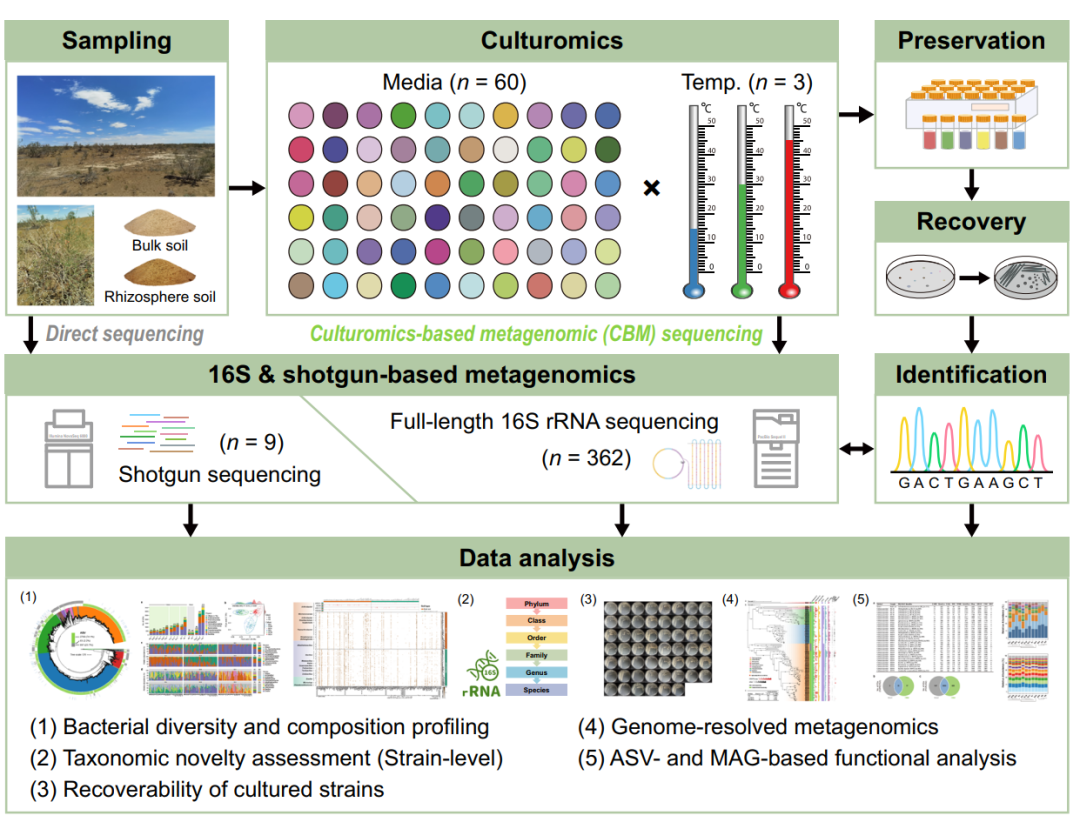
图1. 实验设计示意图。
通过预实验选取了裸土和沙拐枣(Calligonum leucocladum)的根际土壤(n = 2),然后将它们接种到60种不同的培养基上,分别在15、30和45℃下培养,其中在6个亚组中共产生360个培养物:BCL(15 ℃,裸土培养物)、BCM(30 ℃,裸土培养物)、BCH(45 ℃,裸土培养物)、RCL(15 ℃,根际土培养物)、RCM(30 ℃,根际土培养物)和 RCH(45 ℃,根际土培养物)。对2个原始土壤和360个培养富集物样品进行了PacBio SMRT全长16S rRNA基因测序。此外,还对2份原始土壤样本和7份选定的培养富集物进行了鸟枪法宏基因组测序。每份培养富集物保存在-80 ℃的甘油(25%,v/v)中,其中2份用于二次恢复性分离验证(BM11和RM11分别为裸土和根际土使用M11琼脂培养基在30 ℃下培养所得培养物)。
- 结果 -
① CBM捕获了在直接测序中被遗漏的大量ASVs
CBM captures the majority of ASVs missed by direct sequencing
通过全长16S rRNA基因测序,共获得 4,610,948个CCSs,其中2份原始土壤样品分别有12,913(Bulk:裸土)和12,980(Rhizosphere:根际土)个,360个培养富集物样品从8,213到13,220不等。经过barcode识别、质量过滤、去噪和去除嵌合体,共得到3,666,324个有效的CCSs。原始裸土和根际土分别包含6,299和6,328个有效CCSs。而培养富集物平均包含10,121个有效CCSs,从6,354个到11,778个不等。362个样本共产生了3,779种不同的ASVs。
基于全长16S rRNA基因测序的CBM策略比不依赖培养的直接测序回收了更多数量的ASVs(图2a)。直接测序共回收了980个ASVs,其中83个被CBM捕获,而CBM还检测到了另外2,799个ASVs(图2a)。此外,在每个培养富集物中平均检测到49个ASVs。同时,我们还对从门到种不同分类水平进行了分析,结果表明,在属水平,仅被CBM检测到的有142个属(仅直接测序检测到有102个属),CBM和直接测序均检测到的有63个属。同样地,在门水平,CBM检测到了3个独特门类(Spirochaetota、Fusobacteriota和Synergistota),它们在直接测序中没有被发现。相比之下,有9个门类仅通过直接测序发现,分别为Gemmatimonadota、Nitrospirota、Planctomycetota、Acidobacteriota、Chloroflexota、Armatimonadota、Theromicrobiota、BRC1 和 Rhodothermota(图2b)。总之,这些结果表明,基于全长16S rRNA基因扩增子测序的CBM策略,我们可以从沙漠土壤中获得相当丰富的ASVs和物种分类多样性,这极大地填补了直接测序所遗漏的绝大多数。
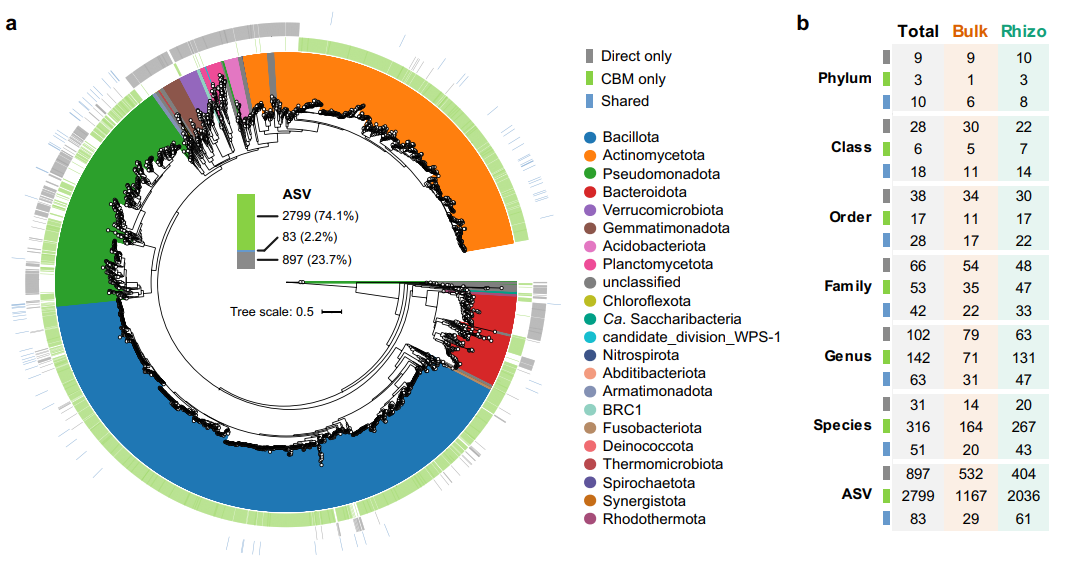
图2. 直接测序和CBM策略恢复的ASV数量与和分类比较。
a 整个数据集中分别有74.1%(2,799个)和 23.7%(897个)的ASVs仅在直接测序(灰色)和CBM(绿色)中检测到。
b 通过直接测序和CBM在不同分类级别上检测到的细菌分类群的数量。Bulk,裸土;Rhizo,根际土。
② 原始土壤及其相关培养物的细菌多样性与分类组成
Bacterial biodiversity and taxonomic profiles of original soils and associated cultures
所有样品的稀释曲线均接近饱和,表明每个样本的微生物多样性均得到了充分覆盖(图3a)。等级丰度曲线表明,原始土壤的丰富度和均匀度远高于任何相关的培养物样品(图3b)。此外,我们还发现2种原始土壤之间以及3种培养温度之间的细菌群落存在显著差异。α多样性指数也表明,每个培养富集物的多样性均明显低于相应的原始土壤样品。此外,对不同亚组下的ASV总数进行了统计,并在门水平进行了分配(图4a)。
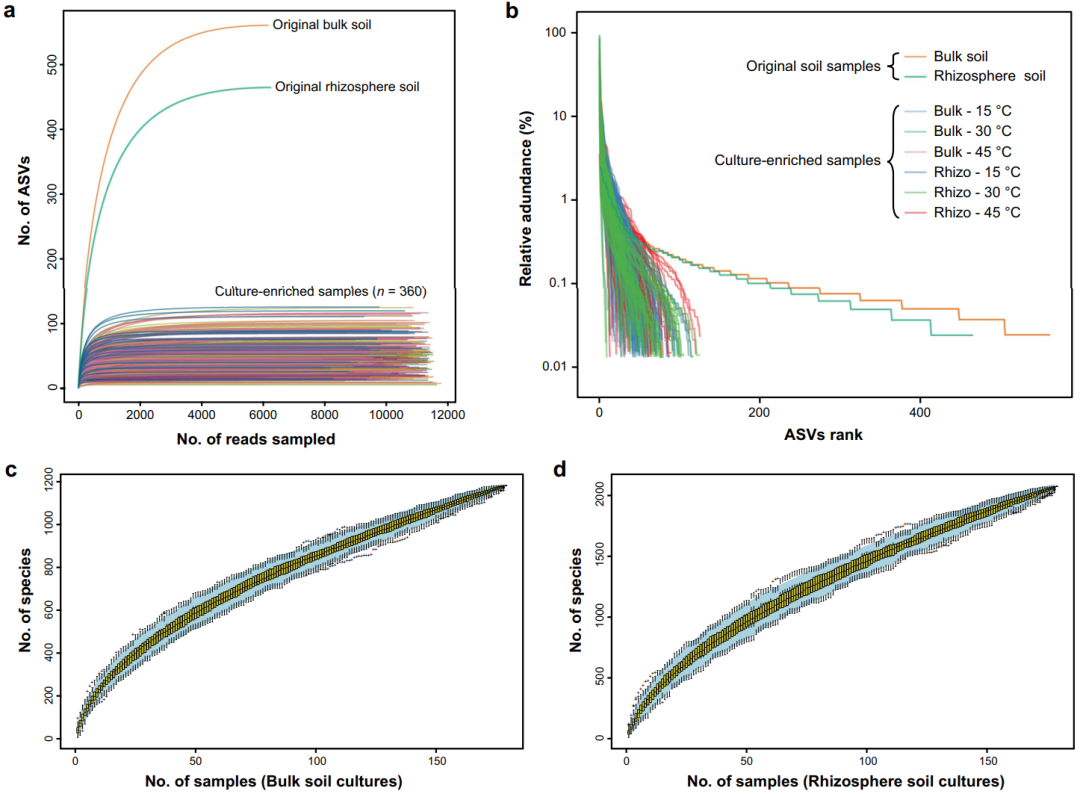
图3. 所有样本的稀释曲线、等级丰度曲线和物种积累曲线。
a 稀释曲线。
b 等级丰度曲线。
c, d 物种积累曲线。裸土和根际土培养物的物种累计曲线均进行1000次随机取样,置信区间为95%。
不同的培养条件对捕获细菌群落的多样性起着重要作用。图4b,c和d显示了在不同培养基和温度条件下,通过直接测序和培养富集全长16S扩增子测序所反映的细菌群落分类分布以及β多样性关系。结果显示,使用选择性培养基和不同的培养温度均促进了细菌分类群的发展(培养组学产生的所有平板图像见补充图像数据集,n = 1,800)。此外,PERMANOVA分析结果表明,培养群落受温度、培养基和土壤的显著影响(p = 0.001),分别可解释总细菌群落变异的20.5%、19.8%和6.3%。为进一步明确不同培养条件下的细菌类群,进行了LEfSe分析,结果显示不同处理对不同类群的组成和多样性具有显著影响。这些结果表明,沙漠土壤中培养的细菌类群会受到不同温度、培养基和土壤特性条件的剧烈影响,我们需要进一步去认识这些尚未被探索的类群的代谢功能。
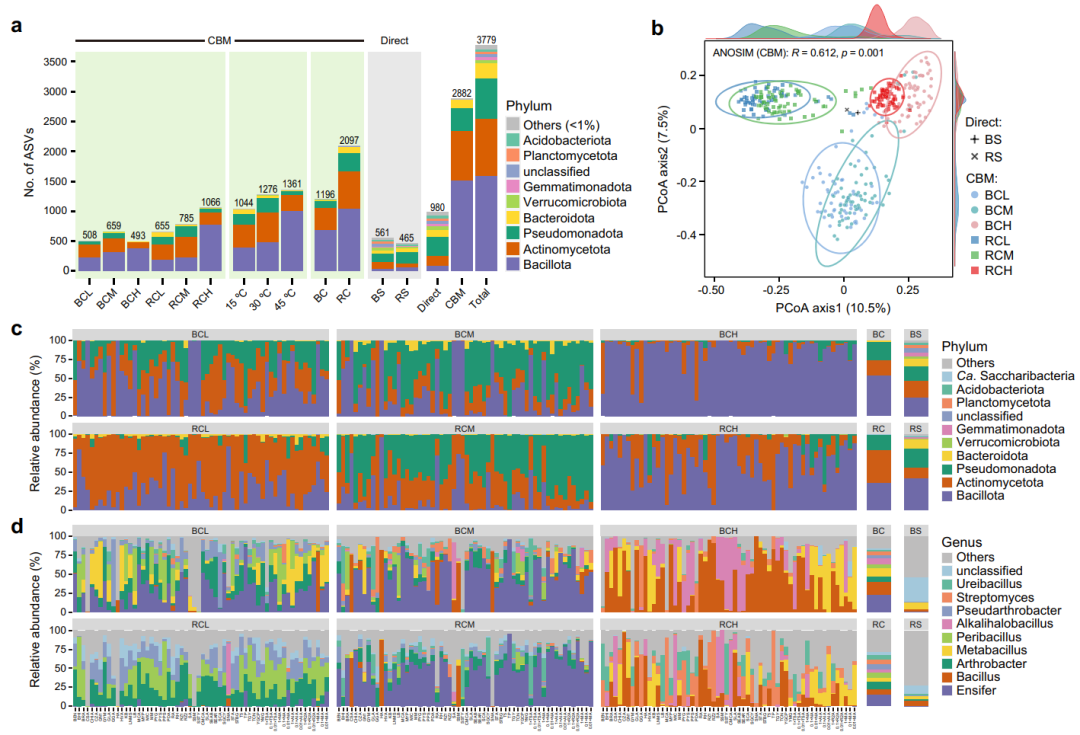
图4. 原始土壤及其相关培养物样品的细菌多样性及组成。
a 原始土壤的直接测序和不同亚组下培养物的CBM策略检测到的ASV数量(标有门级分类和着色)。
b 基于binary Jaccard距离的PCoA图显示了不同样品间的细菌群落相似性。
c, d 柱状图分别描述了在门和属水平上注释的所有ASVs的分类归属;列出了相对丰度最高的前10个分类群和“Others”。BC,裸土培养物;RC,根际土培养物;BS,原始裸土;RS,原始根际土;其它缩写同图 1。
在门水平,2份原始土壤样品以Bacillota、Actinomycetota、Pseudomonadota和Bacteroidota为主,在整个培养富集样品中这4个门也占绝对优势(图4c)。在属水平,我们发现培养富集物样品的6个亚组(土壤-温度)差异十分明显,直接测序检测到的丰富属与CBM存在显著不同(图4d)。同样地,图5显示了通过CBM策略获得的204个属的平均相对丰度(包括“未分类”的属)。结果表明,使用2种土壤和不同的培养基可培养出多种多样的细菌属。此外,我们也统计了培养富集样本中含量最高的35个ASVs(>0.5%)的相对丰度。相对丰度最高的ASV1被归类为Ensifermeliloti(同义词:Sinorhizobium meliloti),几乎所有培养基(CSA和SA除外)都能回收它。在不同的培养条件下,属于Alkalihalobacillusclausii的4个 ASVs(ASV5、ASV22、ASV23和ASV34)表现出十分相似的分布特征。总之,系统的培养组学实验与全长16S rRNA扩增子测序相结合大大提高了沙漠土壤微生物区系的分类多样性与分辨率。
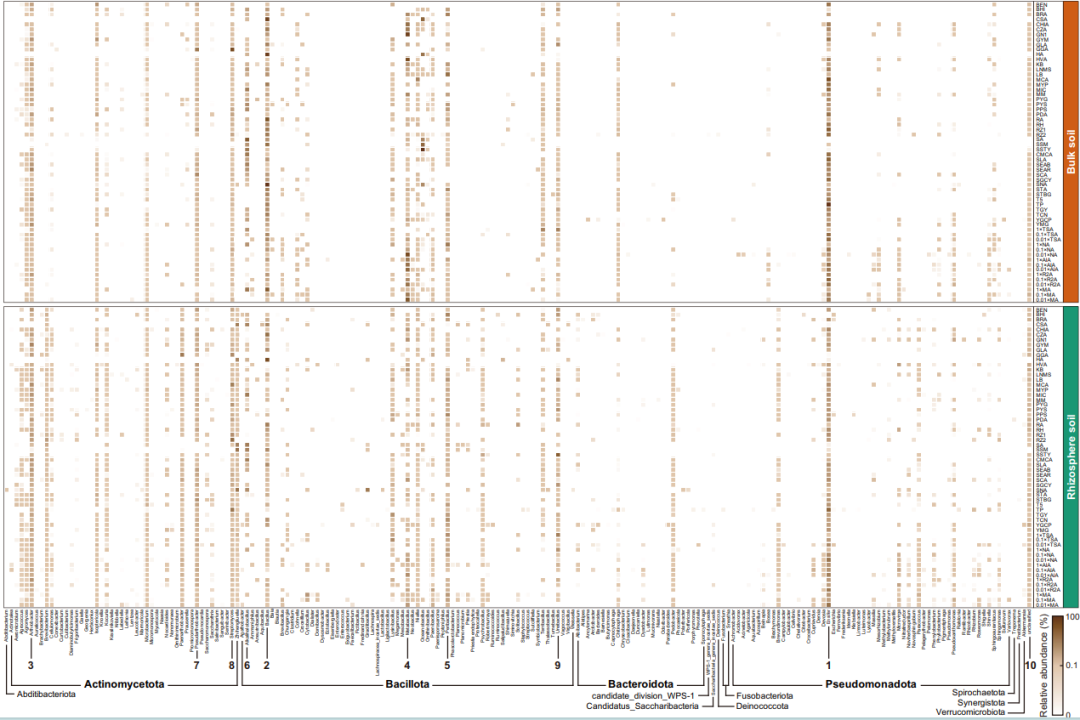
图5.
通过60种培养基获得的裸土(橙色)和根际土(绿色)培养物在不同属水平的平均相对丰度。横坐标表示通过培养组学获得的12个门下的204个属(包括“未分类”的属)(按字母顺序排列)。相对丰度最高的前10个菌属(Ensifer, Bacillus, Arthrobacter, Metabacillus, Peribacillus, Alkalihalobacillus, Pseudarthrobacter, Streptomyces, Ureibacillus和unclassified)被用数字标出(按排名顺序)。
③ 菌株水平分析揭示了沙漠土壤中数量巨大且可培养的细菌新类群
Strain-level analysis reveals a large number of cultivable novel bacterial taxa in desert soils
通过与NCBI 16S rRNA数据库进行批量BLAST搜索,分析了本研究所有3,779个ASVs的序列新颖性。经统计,所有样本(n = 362)中共检测到1,941个ASVs(51.4%)被鉴定为潜在新分类群(Potentially novel taxa,PNT),剩余的1,838个ASVs(48.6%)被归为已知物种(Known species,KS)。如图6a所示,CBM 发现了大量的PNT(1,095个),其中大部分属于潜在新种(1,007个),这远远超过直接测序发现的PNT数量。然而,在所有培养富集样本中,只有88个ASVs被鉴定为属或更高分类等级下的潜在新类群。与此同时,直接测序检测到的PNT以潜在新属为主,有398个ASVs之多,占原始土壤样品总 ASV的40.6%。值得注意的是,有6个16S rRNA基因序列相同度低于75.0%的ASV也被直接测序发现,它们被推断为潜在的新门类群(图6a)。值得注意的是,我们发现只有32个(1.6%)PNT被直接测序和CBM同时检出(图6b)。此外,还分析了培养基对PNT分离培养效果的影响,PNT数量或比例最高的前10种培养基见图6c。这些结果表明,沙漠土壤是一个巨大的细菌新类群宝库,而利用CBM策略则可以发现大量直接测序无法捕获的潜在新物种。
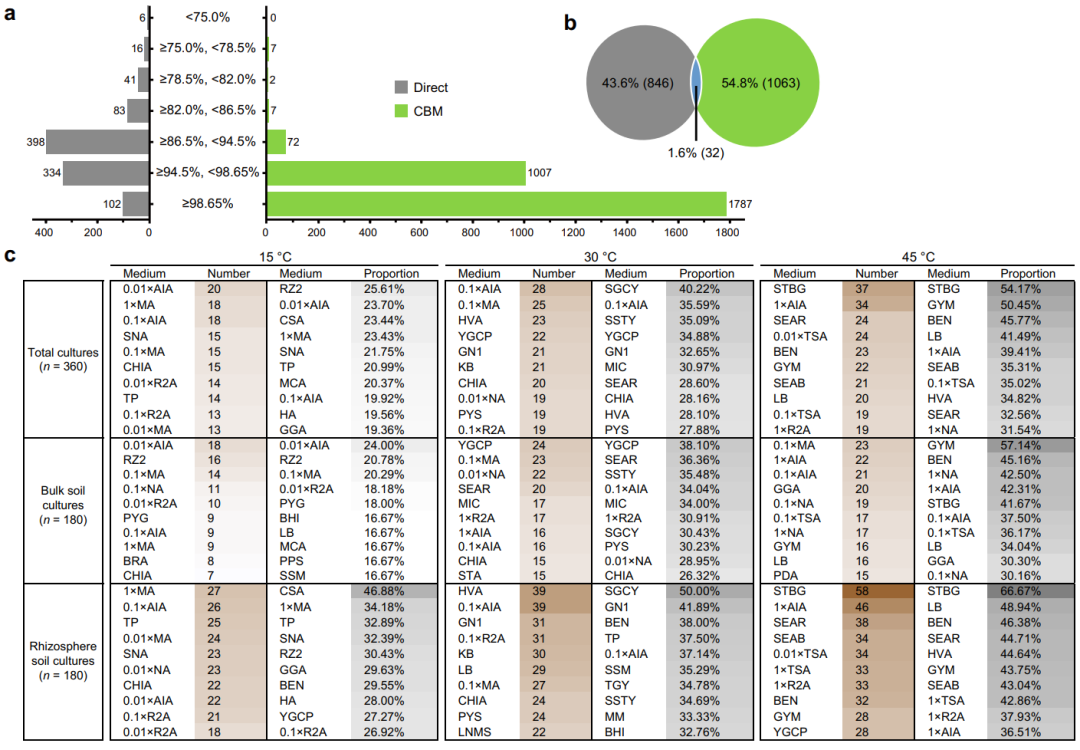
图6. 潜在新分类群概况及相关培养基分离效率排名。
a 直接测序(灰色)和CBM(绿色)检测到属于潜在新分类群的ASV数量,基于不同分类学级别的分类阈值(门:75.0%;纲:78.5%;目 82.0%;科:86.5%;属:94.5%;种:98.65%):
b 韦恩图显示了通过直接测序和CBM策略检测到的特有及共享潜在新类群的ASV数量。
c 潜在新类群数量或比例(%)最高的前10种培养基清单。
④ CBM允许后续恢复感兴趣的微生物
CBM allows for the post hoc recovery of microbes of interest
根据形态学去重复,我们从BM11和 RM11的冷冻细菌培养物中分别分离出26株和 28株细菌(图7a)。这些菌株被分布在7个属(Bacillus、Planococcus、Priestia、Oceanobacillus、Gracilibacillus、Halobacillus和Brevibacterium)。如图7b所示,我们发现有50株菌(占总数的92.6%)的16S rRNA与本研究中的ASV完全匹配。其中,38株与BM11/RM11的ASV完全匹配,12株与非BM11/RM11的ASV完全匹配,其余4株与任何ASV都不能完全匹配。此外,有4株属于芽孢杆菌门(Gracilibacillus sp. RM11-1)和放线菌门(Brevibacterium sp. RM11-19,Brevibacterium sp. RM11-23和Brevibacterium sp. RM11-26)的菌株属于潜在新种(16S rRNA最高相似度小于98.65%)。值得注意的是,候选新种Brevibacterium sp. RM11-19与ASV20的16S rRNA基因序列能够100%匹配,其在培养物RM11中的相对丰度极低,仅0.03%(3个CCSs)。所有菌株的分类情况见附表。这些证据表明,通过CBM策略和测序后二次分离的方法,可以有针对性地分离出感兴趣的微生物。
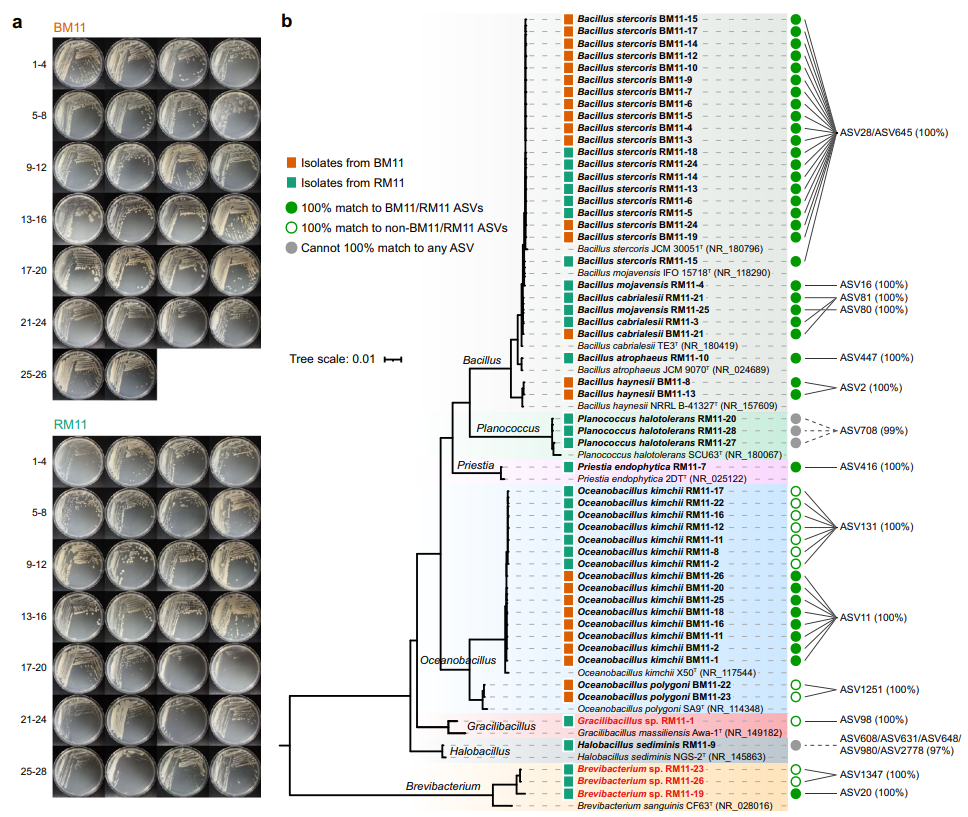
图7. 恢复分离菌株的系统发育树及培养特征。
a 在M11(HA)琼脂上培养5天后,对从BM11收获的26株菌株和从RM11收获的28株菌株进行拍照记录。
b 基于54株回收菌株及其最相似模式物种的16S rRNA基因序列构建的系统发育树(最大似然法)。大肠杆菌ATCC 11775T (X80725)被用作外群。彩色方块表示菌株的分离源,即BM11(橙色)或RM11(绿色)。绿色实心圆表示该菌株能与相应原始培养样本的ASV 100%匹配;绿色空心圆圈表示该菌株能与非BM11和非RM11培养物的ASV 100%匹配;灰色实心圆圈表示该菌株不能与本研究中产生的任何ASV 100%匹配。带有红色和粗体名称的菌株被鉴定为潜在新物种,而其它菌株则被归为已知物种。通过本地blast检测到的最相近的ASV均已列出在图形右侧,括号内百分数为相应的16S rRNA基因序列一致性。
⑤ CBM策略极大提高了MAG的获取率、组装质量和群落功能分辨率
CBM greatly improves the harvestability, assembly quality of MAGs and the community functional resolution
对2个原始土壤和7个培养物样品进行鸟枪法宏基因组测序,共获得原始数据300.14 Gbp,进一步组装产生了790,432个长度≥1,500 bp的contigs。其中,长度≥10 Kbp的contigs有32,515个,长度≥100 Kbp 的contigs有708个,进一步通过binning共得到580个bins,其中包括33个高质量MAGs(HQ MAG)和115个中等质量MAGs(MQ MAG)。利用GTDB数据库进行分类注释,我们发现148个MAGs被归为2个古菌物种和146个细菌物种(图8a, b)。值得注意的是,有32个HQ MAGs来自于根际土培养物,而通过对原始根际土壤进行直接的鸟枪法测序未获得任何HQ MAGs(图8a, b)。其中,2个HQ MAGs(RM44_bin.034和RM56_bin.042)属于Ensifermeliloti,而Ensifermeliloti是我们PacBio SMRT三代16S全长测序数据中相对丰度最高的物种。此外,我们还发现长度≥100 Kbp的contigs的数量与所获得HQ MAGs的数量呈显著正相关(R = 0.95,p = 9e-5)。
通过CBM策略增加的分类多样性直接转化为群落功能多样性的扩充。通过KEGG、COG、antiSMASH等数据库进行注释,得出了每个HQ MAG编码蛋白质的功能。如图9a所示,基于培养富集的鸟枪法测序显然提供了更多的功能识别。例如,RH52_bin.036(Streptomyces sp.)和RH59_bin.028(Saccharothrix sp.)仅从培养物样品中得到,二者基因组中预测到大量的BGCs(图9a)。值得注意的是,这些基于MAGs的功能多样性景观仅来自360个培养物中的7个。CBM策略的另一大优势是,在不同培养条件下获得的MAGs在分类学上有相当大的差异(图8),这可以极大地补充原始土壤直接测序所无法提供的功能注释空白(图9a)。
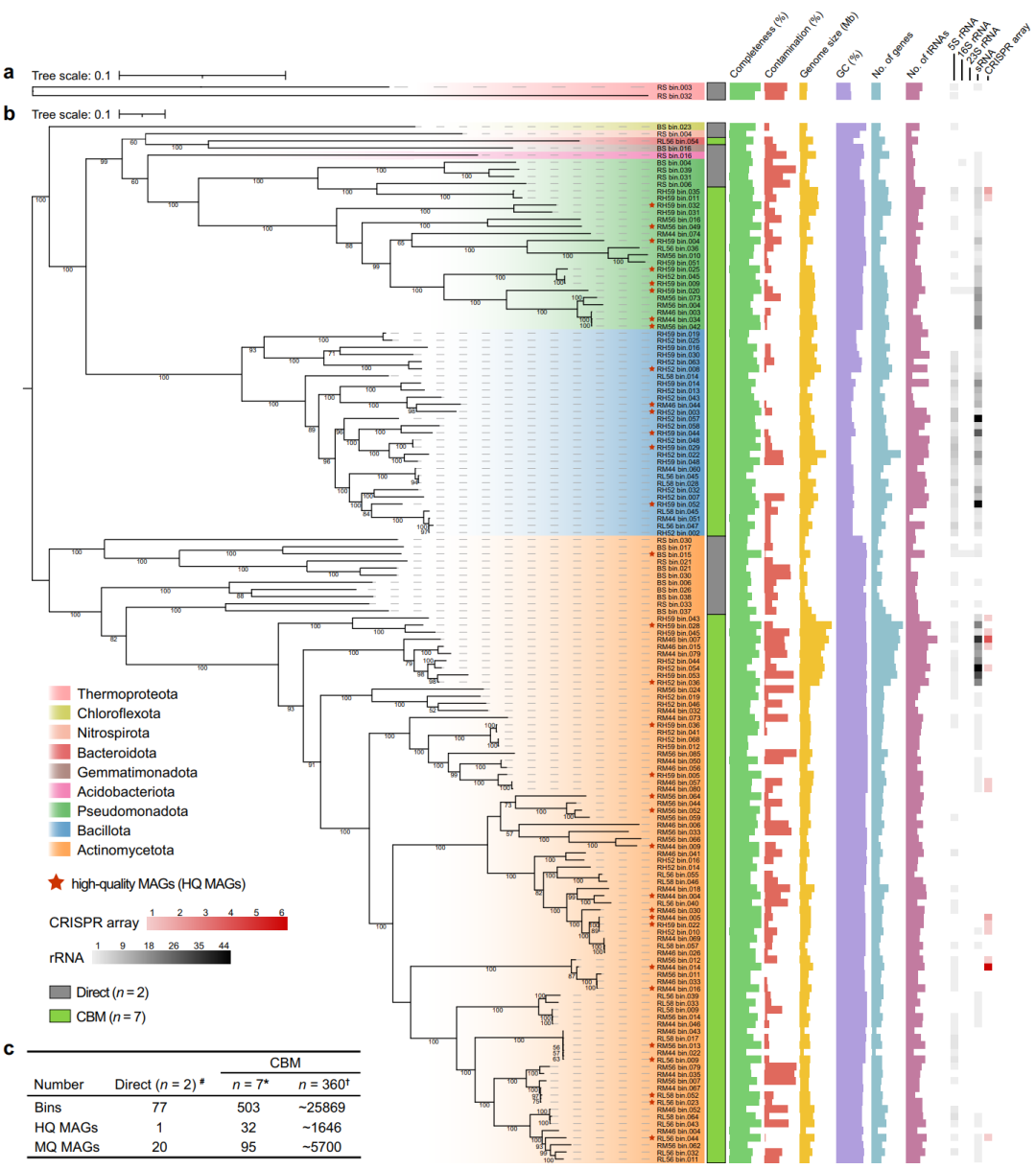
图8. 148个高质量或中质量MAGs的系统发育树及概况。
a, b 两个古菌(a)和146个细菌(b)MAGs的系统发育树。红色五角星表示HQ MAGs(完整度大于90%,且污染度小于5%)。所有MAGs在门级别的分类以不同的背景颜色显示。c 原始土壤(直接测序,n = 2)和培养物(CBM,n = 7)宏基因组样品的组装结果。“#”表示直接测序的样品(裸土和根际土);“*”表示培养富集宏基因组测序的培养物样品(RL56、RL58、RM44、RM46、RM56、RH52和RH59);“†”表示根据7个选定培养物的组装结果对所有培养富集物样品的估值。
相应地,基于全长16S rRNA基因测序数据,利用FAPROTAX和PICRUSt2预测了微生物群落的功能代谢潜力。根据FAPROTAX的结果,在3,779个ASVs中,有1,496个(39.6%)被归入55个功能组(Functional groups,FGs)中的至少一个。其中,通过直接测序预测到38个(69.1%)FGs,由207个ASVs贡献;通过CBM测序预测到52个(94.5%)FGs,由1,326个ASVs贡献。17个FGs是培养物特有的,而只有3个FGs是原始土壤特有的(图9b)。在培养富集物和原始土壤样品中,化能异养作用、需氧化学异养作用和硝酸盐还原作用是相对丰度最高的3种FGs。根据PICRUSt2的结果,共记录了6,811个预测的KOs。其中,5855个KOs为原始土壤和培养富集物样品所共有,147个KOs为原始土壤特有,809个KOs为培养富集物特有(图9c)。事实上,FGs的数量与KOs的数量之间存在极为显著的正相关性(R = 0.71,p <2.2e-16)。
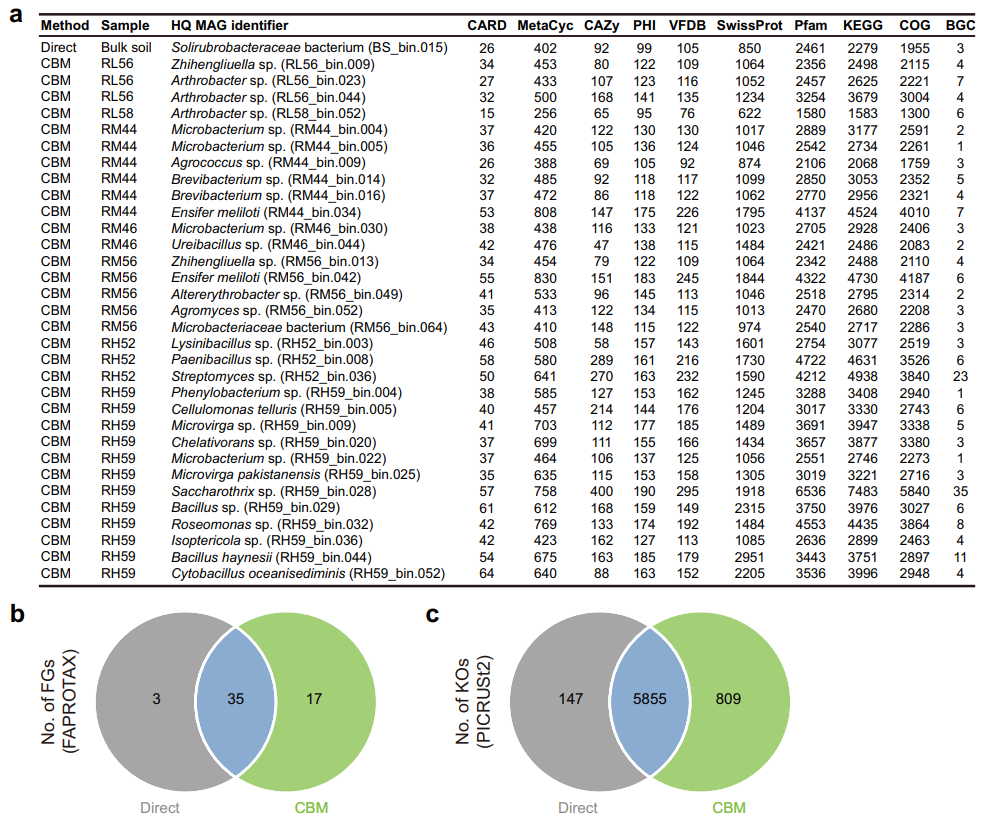
图9. CBM与直接宏基因组测序检测到的功能多样性。
a 所有HQ MAGs(n = 33)的分类及功能概况。列出了每个HQ MAG注释到CARD、MetaCyc、CAZy、PHI、VFDB、SwissProt、Pfam、KEGG、COG和BGC的预测基因数。
b, c 基于直接测序和CBM预测的共有/特有功能组和KEGG直系同源系统的维恩图。用于FAPROTAX/PICRUSt2预测的数据来源于原始土壤和培养物的全长16S rRNA基因测序。FGs,功能组;KOs,KEGG直系同源系统。
- 讨论 -
荒漠(包含沙漠)是地球上最重要的生态系统之一,但由于其极端特异性和缺少生机的特征,一直未受到微生物学家的关注。随着对包括沙漠在内的许多尚未充分开发的极端生境研究的深入和测序数据的积累,这些生境中蕴藏的巨大微生物多样性逐渐被揭示。然而,由于沙漠土壤中细菌群落的高复杂性和低生物量特征,通过直接的宏基因组测序通常难以捕捉到环境库中通常稀有但重要的微生物。此外,以高通量MALDI-TOF的菌落鉴定技术为基础的标准培养物组学方法面临无法完全获取所有培养物信息的问题。在本研究中,我们采用了一种较为系统的培养物组学方法,并提议使用基于培养组的宏基因组学(CBM)策略来放大不同培养条件下选择性富集的特定类群的信号,从而大大简化宏基因组样本的微生物多样性(图1),正如最近在肠道、肺部、废水和沉积物样品等领域中的应用。我们的结果表明,整合大规模培养组学、全长16S rRNA基因扩增子和鸟枪法宏基因组测序的CBM策略可以极大地提高分类学和功能分辨率,从而揭示沙漠土壤中尚未发现的微生物新资源。
利用CBM策略,我们恢复了大部分在免培养直接测序中遗漏的ASVs和微生物多样性(图2),这与之前在各种环境中使用“培养富集宏基因组学”的研究结果一致。这些结果表明,CBM可以成为微生物多样性调查的强大助推器,因为它可以探索低生物量样本中的稀有生物圈,而直接测序通常检测不到或很难检测到这些微生物类群。然而,这并不是要贬低直接测序方法,免培养宏基因组学的系统发育覆盖率往往总是优于培养组学。将免培养技术与微生物培养组学相结合是一种有价值的补充方法,其能让我们更好地了解细菌群落的多样性和不可培养性。此外,我们应意识到全面系统的培养组学方法对于支持CBM策略而言十分重要,因为从单一或少数培养条件中获得的培养富集物样本不足以反映可培养群落的绝大部分特征。在本文中,虽然我们对每个土壤样本均使用了相对全面的培养条件(n = 180),但物种积累曲线仍然是不饱和的(图3c, d),因此,增加新的培养条件势必会获得额外的微生物多样性。
培养物组学是分离未被充分研究的新微生物的有效策略,而后者则是挖掘环境微生物资源的绝佳材料。基于直接测序和CBM策略产生的全长16S序列数据的菌株水平分析结果,我们发现沙漠土壤绝对是一个巨大的未知细菌新资源的宝库。令人鼓舞的是,超过半数的ASVs(1941个,占比51.4%)总体上可归类为潜在细菌新分类单元,其中1095个是可培养的。然而,尽管使用了全面的多重条件,培养高级分类单元的细菌新类群似乎仍然非常困难,直接测序检测到的大量属于高级分类单元的新分类群证明了这一点(图6a)。我们这可能是由于这部分细菌大多对营养要求很低,而低营养类群通常不太适合非原位的培养研究。
目前,可用的微生物纯培养物仍是微生物生理学中深入研究基因、蛋白和代谢途径作用的最重要基石。如图7所示,恢复分离验证结果表明,培养物中感兴趣的微生物(如新物种或特定类群)可能在培养物富集测序后被成功回收。这些结果进一步说明,测序研究可促使有针对性的培养物组学研究培养出感兴趣的微生物,强调了培养物组学研究与免培养研究之间的互补性。事实上,迅速发展的免培养宏基因组研究产生了许多相关性调查结果和未经验证的假说。因此说,从环境微生物群落中分离关键菌株至关重要,而CBM策略允许进行这种分离尝试。
在本研究中,培养富集宏基因组学在高质量基因组重构方面的表现也优于直接宏基因组测序,这反映在MAGs的可用性上(包括多样性、数量和质量水平等方面)(图8)。此外,使用培养富集宏基因组测序方法仅从1.94%的培养物中获得了32个高质量MAGs(HQ MAGs)。相比之下,直接宏基因组测序在实现上述目标方面要逊色得多。此外,即使在每个样本超过30 Gb的高测序深度下,我们也未从根际土壤宏基因组数据中回收任何高质量的MAGs,而从裸土宏基因组数据也仅回收了1个HQ MAG。不过值得注意的是,与培养富集宏基因组相比,直接测序在获得一些未培养微生物的深分支中等质量MAGs方面仍有显著优势,其中包括2个古菌的MAGs(如RS_bin.003和RS_bin.032)(图8a, b)。根据现有培养富集宏基因组测序数据的分析结果,我们估计对全部培养物样品(n = 360)进行测序可能会产生约25,869个bins,1,646个HQ MAGs以及5,700个MQ MAGs(图 8c)。除了提高分类学分辨率外,培养富集宏基因组测序获得的更全面的MAGs(以及 ASVs)恢复也从根本上改变了功能分析的分辨率,这一点在本研究(图9)以及之前其它微生物组研究中也都得到了证实。通过培养富集宏基因组测序和基因组解析宏基因组分析(Genome-resolved metagenomic analysis)扩展了HQ MAGs集,这对深入解析沙漠微生物的功能而言至关重要。有了这些高质量的细菌基因组,我们就能更好地研究微生物暗物质的代谢功能和相关机制,更充分地了解沙漠环境中单个微生物的基因谱系。
然而,这项研究也存在一些局限性。选择性培养条件的应用使低丰度微生物得以增殖,并促进了所研究样本中细菌多样性的恢复。然而根据分析结果,与原始样本的测序结果相比,全长16S扩增子和鸟枪法宏基因组测序的结果同时支持了特定类群的丰度富集,这可能会导致未培养类群被低估或忽略。因此,CBM策略可能无法反映相应环境中微生物组成和功能途径的真实相对丰度。另外,由于本研究只选择了几个有代表性的培养物(1.94%)进行鸟枪法宏基因组测序,因此我们无法完全确定从所有培养物中究竟可以回收多少可用的MAGs。此外,本研究采用的培养组学策略是基于传统的琼脂平板,样本量和培养条件可能仍不足以代表足够的样本异质性和生物多样性。值得期待的是,将CBM策略与一些先进的培养技术(如微流控、极限稀释法和单细胞分选等)和长读数鸟枪法测序技术相结合,可能会克服上述局限性,以更快、更便宜、更方便和更高效的方式实现高通量的细菌多样性探索和基因组重建。我们认为这些改进将使本文提出的基于培养组的宏基因组学研究策略成为更有力的沙漠微生物暗物质挖掘工具。
- 结论 -
将高分辨的基于培养组的宏基因组学(CBM)方法与直接宏基因组测序相结合,能够深入剖析沙漠土壤中的微生物暗物质。作为一种整合了培养组学和宏基因组学(全长16S扩增子和鸟枪法测序)的多组学研究策略,CBM能够极大地提高沙漠土壤微生物组的分类和功能分辨率,且重要的是它能在宏基因组测序结果的指导下对感兴趣的微生物进行恢复性分离。得益于物种水平的分析,研究还揭示了沙漠土壤中尚未被充分开发的细菌新资源的巨大潜力。此外,多重条件组合下的培养组学实验结果为分离沙漠土壤中某些特殊或新的细菌类群也提供了重要参考。有了这些数据,我们能更好地了解沙漠微生物的群落组成和分布、微生物之间的相互作用、环境适应机制以及物种基因库。基于培养组的宏基因组学,为深入认识和挖掘微生物组样本中的暗物质资源提供了一个新的视角,尤其是那些来自于极端或特殊生境的样本。
参考文献
Li, S., Lian, WH., Han, JR. et al. Capturing the microbial dark matter in desert soils using culturomics-based metagenomics and high-resolution analysis. npj Biofilms Microbiomes 9, 67 (2023). https://doi.org/10.1038/s41522-023-00439-8
- 作者简介 -
第一作者

中山大学
李帅
博士后
李帅,2022年博士毕业于中山大学,获理学博士学位,现为中山大学生命科学学院博士后,主要从事荒漠等极端或特殊生境微生物资源的收集、分类、功能挖掘及相关生态学研究。目前以第一作者(含共一)在Science of the Total Environment、npj Biofilms and Microbiomes、International Journal of Systematic and Evolutionary Microbiology、Antonie van Leeuwenhoek和Ecotoxicology and Environmental Safety等期刊发表学术论文16篇。
通讯作者

中山大学
董雷
副研究员
董雷,中山大学生命科学学院副研究员,长期从事极端或特殊生境(沙漠、丹霞地貌和沿海滩涂)微生物分类及系统学、资源评价及应用基础研究。iMeta青年编委,Archives of Microbiology、Current Microbiology等杂志审稿人。先后承担了包括国家自然科学基金委青年,面上项目,科技部第三次新疆综合科学考察项目子课题等多项课题。目前以第一作者或通讯作者(含并列)在Organic Letters、Science of the Total Environment、npj Biofilms and Microbiomes、Environmental Microbiome、International Journal of Systematic and Evolutionary Microbiology等期刊发表学术论文30余篇。

中山大学
李文均
教授,博士生导师
李文均,中山大学生命科学学院百人计划/珠江学者、逸仙学者特聘教授,博士生导师,课题组长,兼任中国科学院新疆生态与地理研究所特聘研究员。长期从事高温、高盐碱和海洋等极端或特殊生境的微生物分类及系统学、生态学研究。现任国际原核微生物系统学委员会(ICSP)国际委员,伯杰氏国际系统微生物学会(BISMiS)创始会员及候任主席。同时兼任中国微生物学会微生物教学工作委员会副主任委员、国际交流工作委员会委员、普通微生物专业委员会委员、地质微生物专业委员会委员、微生物资源专业委员会委员。Microbiome、Environmental Microbiome、Frontiers in Microbiology、International Journal of Systematic and Evolutionary Microbiology、Antonie van Leeuwenhoek、Archives of Microbiology等期刊杂志副主编或编辑,mLife、Systematic and Applied Microbiology、Journal of Arid Land等期刊编委。先后承担并顺利完成国家级或省级课题研究50余项。以通讯/第一作者在National Science Review、Nature Communications、Microbiome、ISME J、Science of the Total Environment、npj Biofilms and Microbiomes等期刊发表学术论文600余篇,参与发表论文400余篇,主编或参编专著10余部。2014-2022连续九年均入选由世界著名出版公司爱思唯尔(Elsevier)发布的中国高被引学者榜单。

















 1090
1090

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









