






药物-微生物相关性能够预测癌症患者的临床结果
High-resolution analyses of associations between medications,microbiome, and mortality in cancer patients

Article,2023-6-08,Cell, [IF 64.5]
DOI:https://doi.org/10.1016/j.cell.2023.05.007
原文链接:https://www.cell.com/cell/fulltext/S0092-8674(23)00526-3
第一作者:Chi L. Nguyen
通讯作者:Marcel R.M. van den Brink
合作作者:Kate A. Markey, Oriana Miltiadous, Anqi Dai, Nicholas Waters, Keimya Sadeghi, Teng Fei, Roni Shouval, Bradford P. Taylor, Chen Liao, John B. Slingerland, Ann E. Slingerland, Annelie G. Clurman, Molly A. Maloy, Lauren Bohannon, Paul A. Giardina, Daniel G. Brereton, Gabriel K. Armijo, Emily Fontana, Ana Gradissimo, Boglarka Gyurkocza, Anthony D. Sung, Nelson J. Chao, Sean M. Devlin, Ying Taur, Sergio A. Giralt, Miguel-Angel Perales, Joao B. Xavier, Eric G. Pamer, Jonathan U. Peled, Antonio L.C. Gomes
主要单位:
郭士纳·斯隆·凯特琳生物医学科学研究生院,纪念斯隆·凯特琳癌症中心 (Gerstner Sloan Kettering Graduate School of Biomedical Sciences, Memorial Sloan Kettering Cancer Center)
斯隆·凯特琳研究所,纪念斯隆·凯特琳癌症中心 (Sloan Kettering Institute, Memorial Sloan Kettering Cancer Center)
成人骨髓移植医学部,纪念斯隆凯特琳癌症中心 (Adult Bone Marrow Transplantation Service, Department of Medicine, Memorial Sloan Kettering Cancer Center)
威尔康奈尔医学院医学系 (Department of Medicine, Weill Cornell Medical College)
纪念斯隆·凯特琳癌症中心儿科 (Department of Pediatrics, Memorial Sloan Kettering Cancer Center)
流行病学和生物统计学系,纪念斯隆凯特琳癌症中心 (Department of Epidemiology and Biostatistics, Memorial Sloan Kettering Cancer Center)
计算和系统生物学项目,纪念斯隆凯特琳癌症中心 (Program for Computational and Systems Biology, Memorial Sloan Kettering Cancer Center)
杜克大学医学中心医学院血液恶性肿瘤和细胞治疗研究室 (Division of Hematologic Malignancies and Cellular Therapy, Department of Medicine, Duke University Medical Center)
纪念斯隆凯特琳癌症中心传染病医学部 (Infectious Disease Service, Department of Medicine, Memorial Sloan Kettering Cancer Center)
芝加哥大学Duchossois家庭研究所 (Duchossois Family Institute, University of Chicago,)
- 摘要 -
识别药物暴露对癌症患者肠道微生物的影响是比较困难的。本研究通过开发和应用一个新的算法(PARADIGM, parameters associated with dynamics of gut microbiota, 肠道微生物动态变化与选定参数的相关性)解析了药物暴露和微生物组成之间的关系。所用的样品来自于一批系统性纵向收集的粪便微生物材料,这些材料是来自于造血干细胞异体移植患者,同时这些材料也具有医疗机构提供的关于患者信息的详细记录。我们发现一些非抗生素药物(包括泻药、止吐药、阿片类药物)与肠球菌属(Enterococcus)相对丰富度的升高和α多样性的降低有关。宏基因组测序数据分析结果进一步表明亚种竞争导致了患者在造血干细胞异体移植(allo-HCT)期间优势菌株的遗传趋同增加,同时发现这与患者抗生素药物暴露显著相关。我们整合了基于药物暴露的两个验证数据集的药物-微生物的相关性分析结果来预测临床结果,结果表明PARADIGM这一方法能够得出一些生物学和临床相关的关于药物暴露如何扰乱或维持患者的肠道微生物组成的结果。
应用PARADIGM算法对大量癌症患者粪便样本的分析结合详细的每日医疗记录揭示了药物暴露和肠道微生物之间的相关性,这些肠道微生物群落与药物暴露的相关性概括了体外研究的结果,同时也对临床治疗结果具有预测性。
- 引言 -
肠道微生物的动态变化被发现与多种疾病有关,并且肠道微生物的动态变化常常与环境暴露包括抗生素的使用和营养物质的缺乏相联系。非抗生素类药物也能造成肠道微生物变化,但是它们在人类中的作用被研究的不够深入,同时由于缺乏可靠的药物暴露数据(例如,基于回顾式的慢性药物习惯性使用调查)以及缺少集中纵向收集的粪便样本,使得研究非抗生素类药物对肠道微生物变化的作用比较困难。另外,一些先前对药物暴露和微生物组成的研究主要集中于一些相对健康稳定的志愿者群体。而造血干细胞异体移植(allo-HCT)的患者在整个治疗过程中表现出较大的粪便微生物组成的变化,这种变化被发现与患者死亡风险升高相关。这些患者在长时间住院治疗过程中使用了多种多样的药物,这一期间有大量的数据在日常的治疗过程中作为他们电子医疗记录的一部分被收集下来。基于这样一个背景,针对这一患者群体,我们整合收集了大量患者的粪便样本,这些样本提供了绝佳的机会来研究肠道微生物对体内药物暴露的响应。
已有的对这些患者的研究主要集中在抗生素对肠道微生物的影响上;然而,许多日常治疗过程中使用的非抗生素类药物在体外实验中展现出抑制微生物活动的现象。此外,造血干细胞异体移植患者肠道微生物的动态变化被观察到要早于使用广谱抗生素药物之前,表明移植相关的癌症治疗和支持护理类药物的使用可能在其中发挥着作用。我们开发、应用以及验证了一个新的计算方法PARADIGM (parameters associated with dynamics of gut microbiota, 肠道微生物动态变化与选定参数的相关性)来分析从造血干细胞异体移植(allo-HCT)患者群体系统收集得到的粪便样本的16S rRNA扩增子和宏基因组测序数据, 研究了药物、肠道微生物组成和临床结果之间的关系。
- 结果 -
聚类分析得到造血干细胞异体移植期间患者肠道微生物组随时间的变化
Clustering captures the temporal dynamics of the intestinal microbiome during allo-HCT
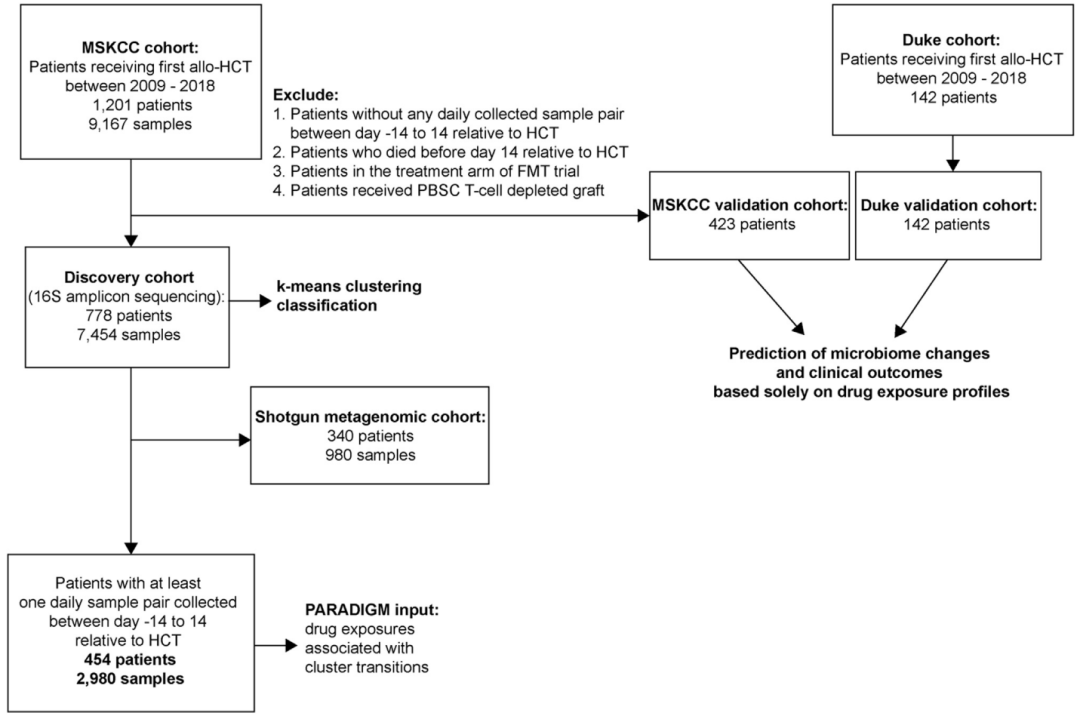
图1. 发现和验证队列的病人选择标准
MSKCC 发现队列被纳入测序数据聚类和 PARADIGM 算法训练集中。验证队列被纳入临床结果的分析中。
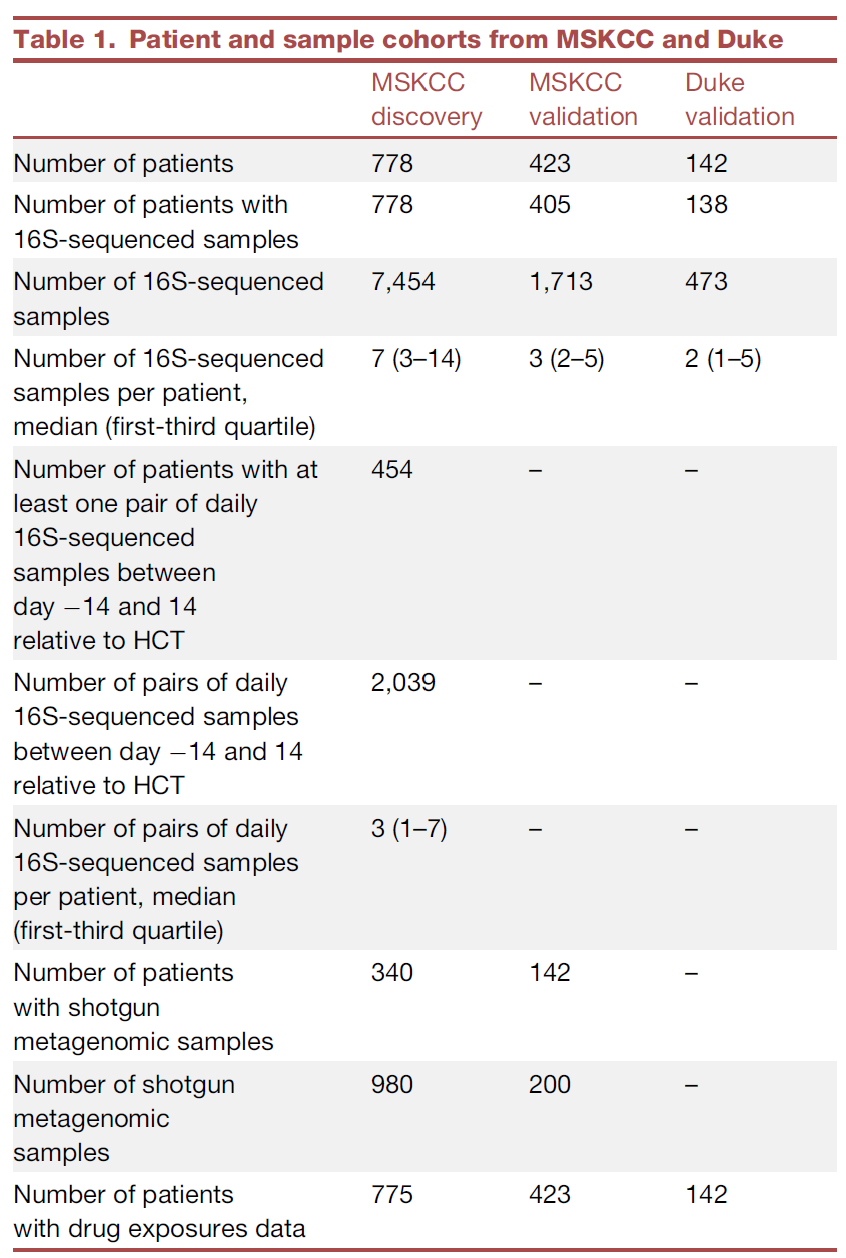
表1. 来自于MSKCC和杜克大学的患者和样本队列情况
数据集包括了来自于纪念斯隆·凯特琳癌症中心(MSKCC)1201名造血干细胞异体移植患者的9167个粪便样本(图 1; 表1)。我们将这些来自于MSKCC的数据分成发现队列(来自于778名患者的7454个样本)和验证队列(来自于423名患者的1713个样本)(图 1; 表1)。利用16S rRNA基因扩增子测序数据结合Bray-Curtis β-多样性差异指数计算了发现队列(discovery cohorts)样本微生物组成在属水平上的差异,利用宏基因组测序样本研究了微生物组成在种水平上的差异,同时利用t-随机邻域嵌入方法(tSNE; t-stochastic neighbor embedding)对高维的粪便微生物组成数据进行可视化(图2A-B)。
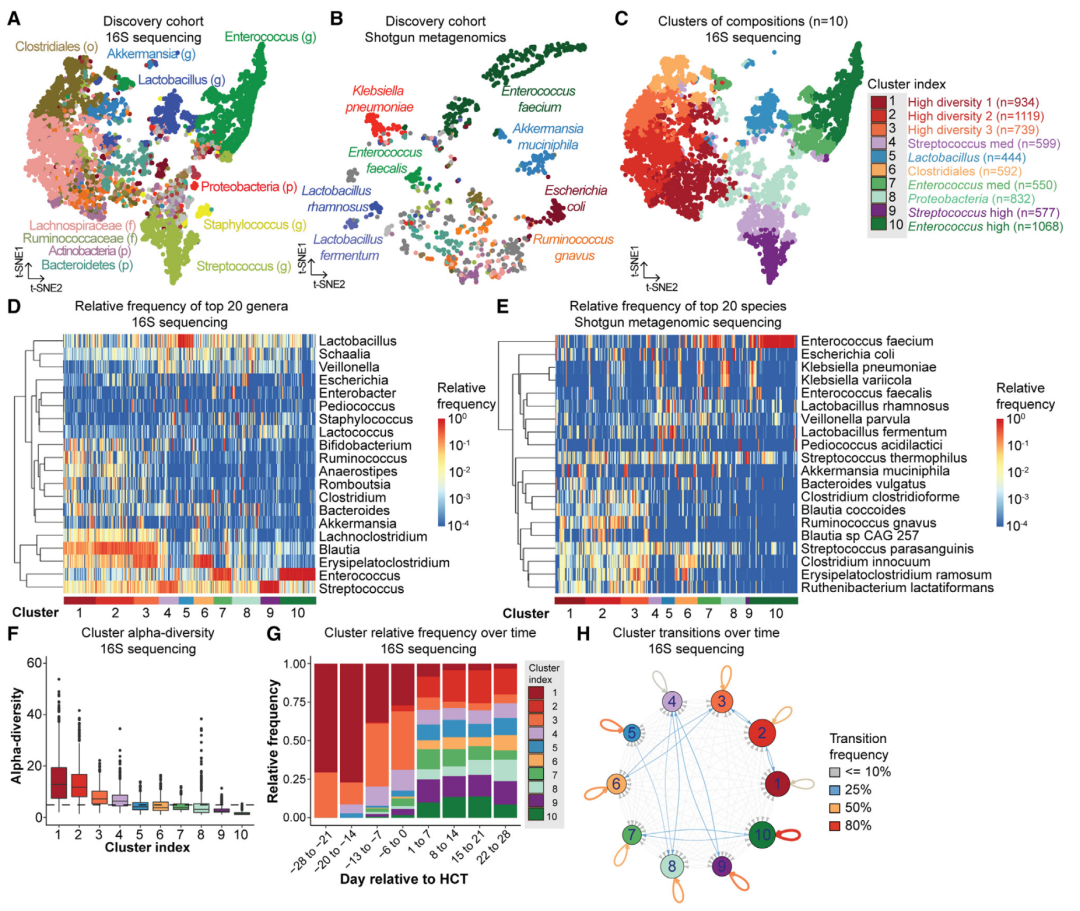
图2. 造血干细胞移植患者的肠道微生物的动态变化。
(A)和(B):在MSKCC发现队列样本中利用tSNE投影得到的肠道微生物群的组成空间图。每一个点代表一个样本,根据相对丰度最高的分类群进行着色,图A是16S rRNA基因分析(7454个样本;778名患者)得到的结果,图B是宏基因组测序(980个样本;340个样本)得到的结果(p代表门,f代表科,o代表目,g代表属)。样品收集的时间是在造血干细胞异体移植前的30天和移植后的2205天之间。
(C):通过k-means无监督聚类分析得到的10个肠道微生物组成的聚类群。
(D)和(E):在MSKCC发现队列样本中相对丰富度最高的20个微生物类群,图D是利用16S rRNA测序数据得到的属水平上的结果,图E是宏基因组测序得到的物种水平上的结果。每一列代表一个样本,每一行代表一个属或种,每行通过分层聚类得到。
(F):聚类群的α多样性(reciprocal Simpson index)。横向短虚线代表MSKCC发现队列样本α多样性的中位数。箱线图代表α多样性的高低和分布。
(G):聚类群的相对频率随着时间的变化。
(H):网络图展示10个肠道微生物聚类群随着时间的相互转换(5482对后续样本;677名患者;采样时间为造血干细胞移植前的16天和移植后的1084天之间)。线条颜色的粗细代表转换频率比例的大小,节点圆圈的大小代表每个聚类群样本数量的大小。
本研究观察到肠道微生物出现损伤,包括α多样性的丧失以及潜在致病微生物群(比如肠球菌属Enterococcus 和肠杆菌属Enterobacteriaceae)的增多(图2A-B)。正如在对这些造血干细胞移植患者描述中指出的一样,这些显著变化的影响可能是深远的,以至于一些单一分类群微生物的丰富度能够占据粪便样本微生物丰富度的90%以上。这一结果对于有害临床结果的预测比如血流感染、移植物抗宿主病(GVHD)和死亡具有预测作用。980个用宏基因组测序的样本分析结果也显示在造血干细胞异体移植期间会导致肠道微生物的损伤(图2B)。特别的,本研究观察到在一组样本中丰富度最高的是各种严格厌氧的细菌(比如活泼瘤胃球菌Ruminococcus gnavus或多枝梭菌Erysipelatoclostridium ramosum),也观察到不同的富含潜在致病性兼性的微生物群落的样本,包括粪肠球菌(Enterococcus faecium)、肺炎克雷伯菌(Klebsiella pneumoniae)和大肠杆菌(Escherichia coli)等。
对比健康人群中更具流动性和非离散型的肠道微生物群落,在造血干细胞异体移植患者中能够重复观察到的这种微生物动态变化提供了独特的理解在环境暴露条件下相对明显的微生物组成或状态紊乱的动态变化和进化的机会。本研究对研究样本的Bray-Curtis β多样性矩阵进行了无监督k-means聚类,并确定了10个不同的微生物聚类群(图2C)。毛螺科细菌(Lachnospiraceae)和梭菌属细菌(Clostridiales)这两类在健康人类肠道中与人类共生的微生物类群在1-3号聚类群中也是常见微生物类群,且1-3号聚类群也具有较高的α多样性(与发现队列中等的α多样性对比),聚类群7-10代表低多样性的“失调”状态(图2D-F)。乳杆菌属(Lactobacillus)、变形菌门(Proteobacteria)和链球菌属(Streptococcus)在聚类群5、8和9中高度富集。聚类群7和10包含肠球菌属(Enterococcus)为优势菌株的样本,聚类群10屎肠球菌(E. faecium)显著富集(图2D-E)。这些微生物聚类群也捕捉到了患者在造血干细胞异体移植期间肠道微生物随时间的动态变化:高多样性聚类群1-3在造血干细胞异体移植之前常见,而低多样性的聚类群尤其是聚类群7-10则在造血干细胞异体移植之后出现的更多(图2G)。
将样本分类成离散的微生物组状态使我们能够利用聚类群转变概率来模拟微生物群落更复杂的变化。本研究观察到在造血干细胞异体移植前16天到移植后1084天之间大部分以7天为间隔收集的连续样本的转变频率。在2987对样本(占比为54.5%)中,患者在两个连续样本中微生物聚类群保持不变(图2H)。患者不太可能在连续样本中保持高多样性聚类群1-3中的类型(平均频率为46.4%;SD为5.0%),对比来看有微生物占主导地位的聚类群比如8-10聚类群则高度稳定(平均频率为65.1%;SD为14.1%)。本研究观察到聚类群的α多样性和自我转变的概率呈现显著的负相关关系,表明高多样性的聚类群相对于低多样性的聚类群来说更不稳定。在2495对样本(占比45.5%)中观察到从一个微生物聚类群转变为另一个微生物聚类群。
我们观察到肠球菌属(Enterococcus)丰富度较高的聚类群10特别的稳定,这一结果十分有趣,因为这个属的微生物和造血干细胞移植后不好的临床结果有关。为了进一步研究肠球菌属(Enterococcus)丰富度高的聚类群稳定性高的驱动因素,我们建立了一个具有lasso罚分(lasso penalty)的逻辑回归模型来分析聚类群10的稳定性与抗生素暴露情况、样本采集时间、α多样性和分类群10中最丰富的前20个属的微生物群落的相对丰度之间的关系。本研究将构建的模型应用到一个在造血干细胞移植前14天和移植后100天连续每天收集的成对样本数据集中,发现葡萄球菌属(Staphylococcus)和丹毒荚膜菌属(Erysipelatoclostridium)相对较高的丰富度和聚类群10的稳定性下降有关。另一方面,相对较高的肠球菌属(Enterococcus)丰富度和聚类群10的稳定性升高有关,表明肠球菌属(Enterococcus)在微生物聚类群中占优势地位导致了支持其自身稳定性的正反馈循环。和预期一致,抗生素药物暴露和聚类群10稳定性升高有关。在本研究中,利用真实的临床数据,我们发现环境因子比如药物暴露和微生物群落的生态相关性能够改变微生物群落的稳定性,特别是在肠球菌属(Enterococcus)富集的微生物群落中。
造血干细胞异体移植期间非抗生素类药物暴露和肠道微生物组组成变化有关
Non-antibiotic exposures during allo-HCT are associated with changes in the intestinal microbiome compositions
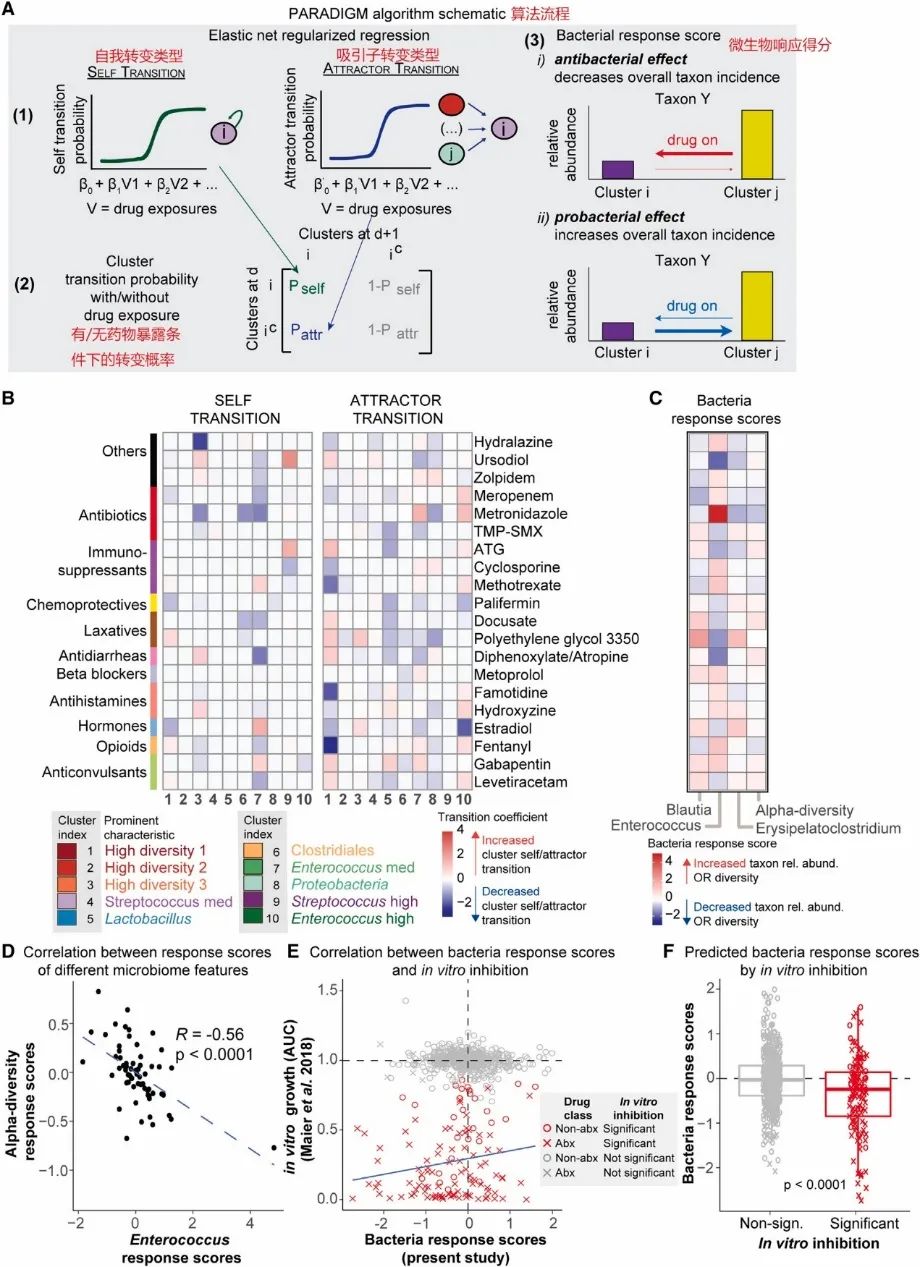
图3. PARADIGM算法预测药物暴露之后微生物组特性比如属水平多样性和α多样性的变化
(A):运用PARADIGM利用每天16S rRNA高通量测序的样本和聚类群转变来推断药物暴露和微生物动态变化之间的相关性的流程示意图。微生物响应得分将药物和微生物聚类群的相关性转变成药物和微生物属的相关性。
(B):药物暴露和微生物组聚类群动态变化之间的相关性。自我转变的系数表示药物暴露是否会增加或减少自然对数转换后的聚类群稳定性。吸引子转变类型的系数表示药物暴露是否会增加或减少对数转换后的转变为给定聚类群的概率。TMP-SMX代表磺胺甲恶唑/甲氧苄氨嘧啶(sulfamethoxazole/trimethoprim);ATG代表抗胸腺细胞球蛋白(anti-thymocyte globulin)。
(C):运用微生物响应得分来预测一个给定的药物暴露和属水平相对丰度的变化或α多样性变化之间的相关性。
(D):肠球菌属(Enterococcus)响应得分和α多样性响应得分之间的皮尔森(Pearson)相关系数。每个点代表一种药物。
(E):微生物响应得分和体外干预的度量值之间的皮尔森(Pearson)相关系数。每一个点代表一个独立的药物物种对之间的相关性。
(F):预测的药物干预下的微生物响应得分。统计检验采用的是双边的Wilcoxon秩和检验。箱线图展示了响应得分的分布。
为了研究药物暴露和微生物聚类群转变之间的相关性,我们开发了PARADIGM算法,这是一个基于逻辑回归模型结合一阶马尔科夫链的计算工具。马尔可夫链模型之前已经被用于研究微生物的动态变化,但是马尔可夫转变和环境因子的相关性并没有被广泛研究。这一模型利用每天收集的粪便样本微生物的16S rRNA高通量测序数据来推断药物暴露和聚类群转变之间的相关性(图3A)。对于每一个聚类群,我们定义了两个转变的类型:自我转变类型(患者肠道微生物组维持在同一个聚类群不变)和吸引子转变类型(患者肠道微生物组转变到一个给定的聚类群)。这两种转变类型的命名主要是考虑其直接的含义能够传达聚类群的动态变化。自我转变类型描述了一个给定的聚类群在每天收集的一对样本中保持其最近状态的概率,而吸引子转变类型则描述了一个给定的聚类群在每天收集的一对样本中从除了其自身的任何聚类群接收转变的概率。
我们研究了62种发现队列患者经常使用的药物对聚类群转变概率的影响。为了研究每一种药物对聚类群自我转变和吸引子转变概率的作用,我们采用弹性网络逻辑回归进行分析,每一种药物分析结果的回归系数可以表示药物和每日聚类群转变相关性的方向和大小。在造血干细胞异体移植期间微生物组损伤的模式和造血干细胞异体移植的时间紧密联系。因此,本研究将时间作为协变量放入模型以解决药物暴露时间模式的影响,并降低微生物组聚类群动态变化中的时间依赖变异。考虑到在测试数据集中每名患者对数据贡献不等的情况,我们在测试组中预先设定了10倍交叉分区验证,这样同一患者的样本就总是出现在同一分区中。
本研究鉴定出一些药物暴露与聚类群自我转变和吸引子转变类型之间的相关性(图3B)。与预期一致,一些过去用于治疗粒细胞减少性发热疾病的药物被发现与肠道微生物的变化相关。美罗培南(meropenem)(增长2.5倍)和甲硝唑(metronidazole)(增长3.4倍)暴露被发现与微生物聚类群向肠球菌属(Enterococcus)丰富度高的聚类群10转变增长有关,这一发现与前人的一些研究相一致。一些非抗生素类药物也与一些具体的聚类群的变化相关。阿瑞匹坦(aprepitant)(一种用于治疗化疗所引起呕吐反应的药物)暴露和向肠球菌属(Enterococcus)丰富度高的聚类群10转变的频率增长2.8倍有关。相似的,镇痛药(阿片类镇痛药芬太尼)暴露和向肠球菌属(Enterococcus)丰富度高的聚类群10转变的频率增长1.9倍有关。其它药物比如拉贝他洛尔(labetalol)和胰岛素(insulin),这些过去不知道会对肠道微生物产生作用的药物被发现与肠球菌属(Enterococcus)丰富度高的聚类群10的稳定性降低有关。总之,运用PARADIGM方法我们发现了一些非抗生素类药物与肠道微生物组变化之间的相关性。
之前的研究已经鉴定出一些具体的微生物,这些微生物与造血干细胞异体移植之后的临床结果具有有利或者有害的关系。基于此,本研究通过计算细菌反应得分,将药物和聚类群的相关性转化为药物和分类单元的相关性,以确定药物暴露与本研究感兴趣的临床相关的特定分类单元变化之间的相关性(图3C)。在我们的模型中,细菌反应得分评估了药物暴露和一个微生物组特征之间的相关性,这些微生物组特征指的是微生物组的分类单元或α多样性,正数反应得分表明药物暴露与较高的相对丰富度或多样性指标有关(图3A)。本研究将研究集中于4个微生物组特征上(之前的研究发现这4个微生物组特征和患者造血干细胞异体移植之后的临床结果有关),包括肠球菌属(Enterococcus)、布劳特氏菌属(Blautia)、丹毒荚膜菌属(Erysipelatoclostridium)的相对丰富度和α多样性。大多数基于经验或者病原靶向使用的抗生素类药物(甲硝唑、美罗培南、氨曲南和头孢吡肟)与肠球菌属(Enterococcus)的相对丰富度增加有关,也与α多样性降低有关,这些发现与以前的研究结果一致(图3C)。头孢吡肟(cefepime)暴露被发现与肠球菌属(Enterococcus)的相对丰富度增加有关,这和之前的研究报道相一致,头孢吡肟的作用可能部分归因于头孢吡肟的活性较差或者存在其它抗肠球菌属(Enterococcus)的头孢菌素。哌拉西林-他唑巴坦(Piperacillin-tazobactam)暴露与肠球菌属(Enterococcus)的相对丰富度降低有关,其相对于其它根据经验使用的抗生素类药物来说也与其它肠道微生物比如布劳特氏菌属(Blautia)、丹毒荚膜菌属(Erysipelatoclostridium)的相对丰富度降低具有更强的相关性。我们也发现和肠球菌属(Enterococcus)的相对丰富度增加强相关的药物是一些非抗生素类的药物,包括阿片类药物比如芬太尼(fentanyl)和氢吗啡酮(hydromorphone)、一些激素类药物比如左旋甲状腺素(levothyroxine)、以及抗惊厥药物比如加巴喷丁(gabapentin)(图3C)。之前的研究发现阿片类药物暴露与没有接受抗生素治疗的ICU病房病人的布劳特氏菌属(Blautia)的相对丰富度降低有关,在本研究中阿片类药物芬太尼(fentanyl)和氢吗啡酮(hydromorphone)也有这种作用。相反的,泻药(laxatives)比如多库酯钠(docusate)和聚乙二醇(polyethylene glycol)被发现与肠球菌属(Enterococcus)的相对丰富度降低具有强相关性(图3C)。之前的实验研究表明聚乙二醇(polyethylene glycol)的使用在全球尺度上导致老鼠的微生物组成发生变化,其作用方式包括调节肠道渗透压或者通过直接的抗菌抑制产生作用。总之,和α多样性维持相关的药物暴露和肠球菌属(Enterococcus)的相对丰富度增长有关,反之亦然(图3D)。
基于真实病例数据集对模型模拟结果进行验证
Validation of in silico findings from real-world patient dataset against an independent in vitro dataset
我们通过使用来自于癌症患者的已发表的真实案例数据集来对比模型模拟的结果,以检验PARADIGM方法的预测能力(图3E-F)。已发表的外部数据集和本研究数据具有19个相同的微生物种并且使用了34种相同的药物。在图的左下象限(图3E),本研究观察到具有体外抑制作用(抗生素和非抗生素)的药物-物种对的富集,对应在模型结果中出现负反应得分(Fisher’s Exact Test: odd ratio = 0.60, p value = 0.01)。此外,与在体外不显示抑制作用的药物相比,显示体外抑制作用的药物-物种对在患者数据集中的反应评分显著降低(图3F)。总之,这些结果显示PARADIGM方法能够准确的预测抗生素类药物和非抗生素类药物的体外抗菌活性,并能够根据在药物暴露情况下对临床症状的潜在影响区分药物和微生物物种的直接相互作用。
抗生素药物暴露是微生物亚种动态变化的强预测因子
Antibiotic exposure is a strong predictor of subspecies dynamics
一些实验研究证实一些药物的杀菌谱是在同一属的物种内特别针对某些物种和株系。因此,我们研究了药物暴露和在造血干细胞异体移植过程中对临床比较重要的属水平菌群的相对丰富度的变化之间的相关性。我们使用从340名患者收集得到的980份样本(MSKCC发现队列数据的一部分)进行宏基因组测序。运用线性混合效应回归模型,我们鉴定出药物暴露和微生物物种相对丰富度变化的相关性。同样的,我们发现不同类型的药物(抗生素、泻药、止泻药和阿片类药物)暴露和类球布劳特氏菌(Blautia coccoides)、延长布劳特氏菌(Blautia producta)、粪肠球菌(Enterococcus faecalis)、屎肠球菌(Enterococcus faecium)和但多枝梭菌(Erysipelatoclostridium ramosum)的相对丰富度变化有关,尽管这些相关性在统计上并不显著。
在人类肠道微生物群落中,大多数物种都以某一单一的主要菌株作为代表,尽管之前在造血干细胞异体移植患者的肠道微生物组处在屎肠球菌(E. faecium)占优势的阶段时观察到复杂的菌株的动态变化。对于我们比较感兴趣的分别属于3个属的5个微生物物种,我们利用StrainPhlAn算法基于标记基因多态性鉴定了优势菌株的序列特征,并且计算了患者连续样本中优势菌株之间系统发育距离(图4A)。连续样本菌株之间较小的系统发育距离表明优势菌株收敛,而较大的系统发育距离表明优势菌株随时间变化逐渐离散。对于大多数微生物物种,特别是屎肠球菌(E. faecium),连续样本之间的系统发育距离随时间推移下降(图4A)。这种随时间的变化模式表明多个物种间的遗传变异在减小,同时一个优势亚型出现。此外,亚型的变异与物种相对丰富度呈负相关关系(图4A)。这种相关性可能是所谓的“选择性扫描”的结果,也就是在造血干细胞异体移植期间通过选择相对适合度更高的菌株或由于种群瓶颈的出现导致物种变异丧失。有研究观察到从造血干细胞异体移植患者系统性收集的粪便样品中分离得到的万古霉素耐药屎肠杆菌(vancomycin-resistant E. faecium)中,其亚种分化出现一种抛物线状的适合度景观类型,这一点是通过优势菌株的菌株分类方法无法得到的。我们观察到抗生素药物暴露在屎肠杆菌(E. faecium)优势菌株的遗传收敛中是一个重要的预测因子,而非抗生素药物则不是(图4B-C)。
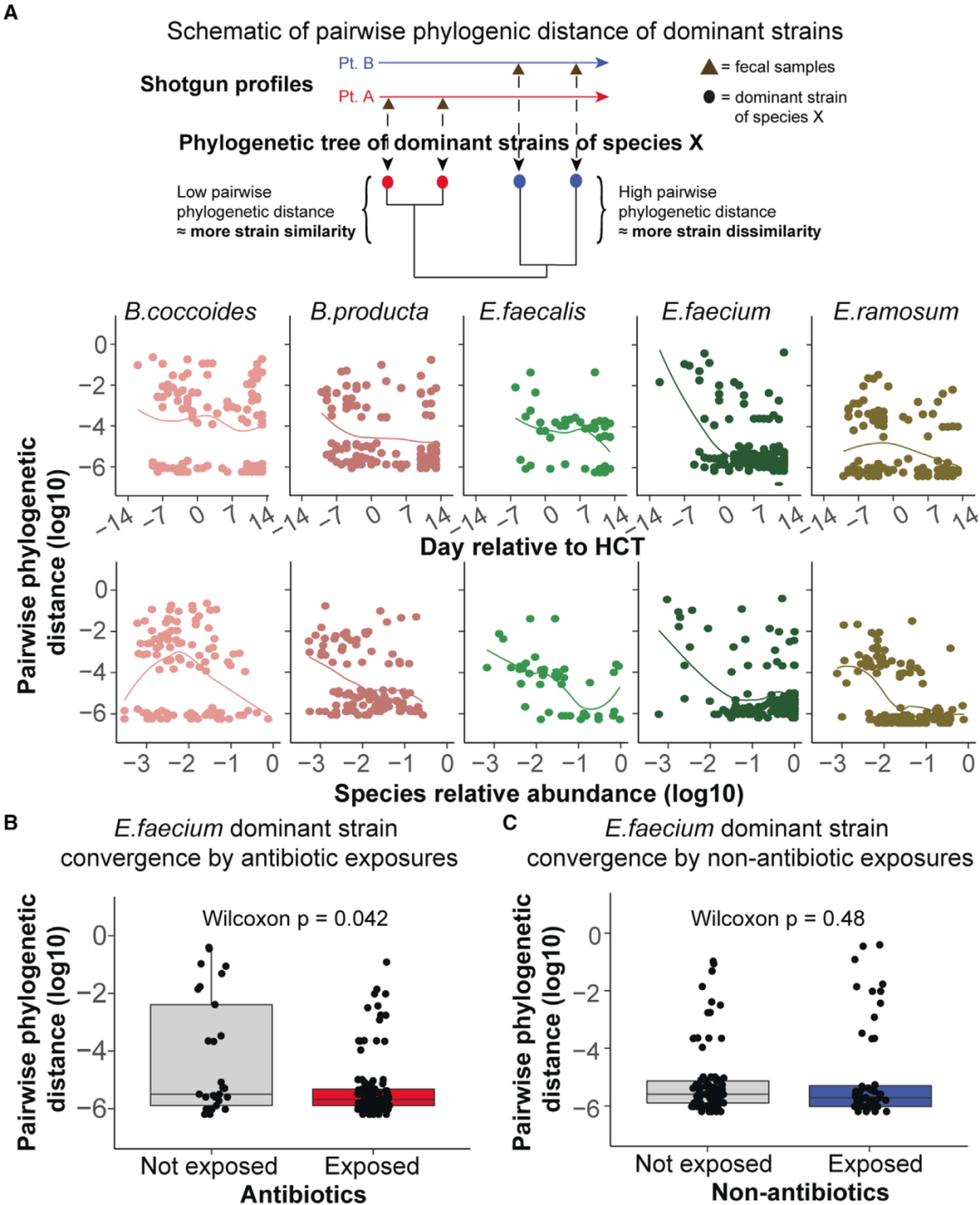
图4. 抗生素类药物是造血干细胞异体移植期间微生物菌株遗传趋同(genetic convergence)的强预测因子
(A):微生物菌株随着时间变化出现遗传趋同(中间行),或者随着物种相对丰富度变化出现遗传趋同(底部行)。每一个点代表连续收集的一对样本中的给定物种的优势菌株之间的系统发育距离。较高的系统发育距离表明遗传差异,而较低的系统发育距离表示菌株遗传的相似性。
(B)和(C):图B是抗生素药物的结果,图C是非抗生素药物的结果。抗生素药物暴露和优势菌株屎肠杆菌(E. faecium)遗传趋同升高有关。每一个点代表优势菌株屎肠杆菌(E. faecium)在一对连续收集样本之间的遗传发育距离,通过每对样品收集之间的时间间隔进行分层。统计检验采用的是双边的Wilcoxon秩和检验。箱线图展示了系统发育距离的分布。
药物和微生物组的相关性对于造血干细胞异体移植之后未来微生物组的变化轨迹和临床治疗结果具有预测性
Drug-microbiome associations are predictive of future microbiome trajectories and clinical outcomes following allo-HCT
基于上文我们研究了药物暴露和微生物变化之间的相关性,并且将粪便微生物组成和造血干细胞异体移植的临床结果相联系。进一步,我们研究了在独立于微生物组数据的情况下,只使用药物暴露的数据是否也能够预测患者的死亡率。运用单独的MSKCC验证组的药物暴露数据,我们定义了患者特定的响应分数指标来量化微生物组特征对药物暴露的净响应(图5A)。例如,患者对于肠球菌属(Enterococcus)特定的响应分数就代表了患者在药物暴露情况下出现肠球菌属(Enterococcus)丰富度增长的相对风险大小(图5B)。我们在两个独立的验证组中检验了患者特定的微生物响应分数与全因死亡率和特定原因死亡率的结果之间的关系。两个独立的验证组中,一个验证组是包含423名来自于MSKCC的患者的数据,这些数据没有包含在PARADIGM方法测试组数据中的;还有一个验证组是包含142名来自于杜克大学医疗中心的独立患者组。在MSKCC验证组中考虑了所有62种药物暴露的情况,而在杜克大学医疗中心的独立患者验证组中只考虑了抗生素类药物暴露的情况。两个队列患者的特征描述参见表2。
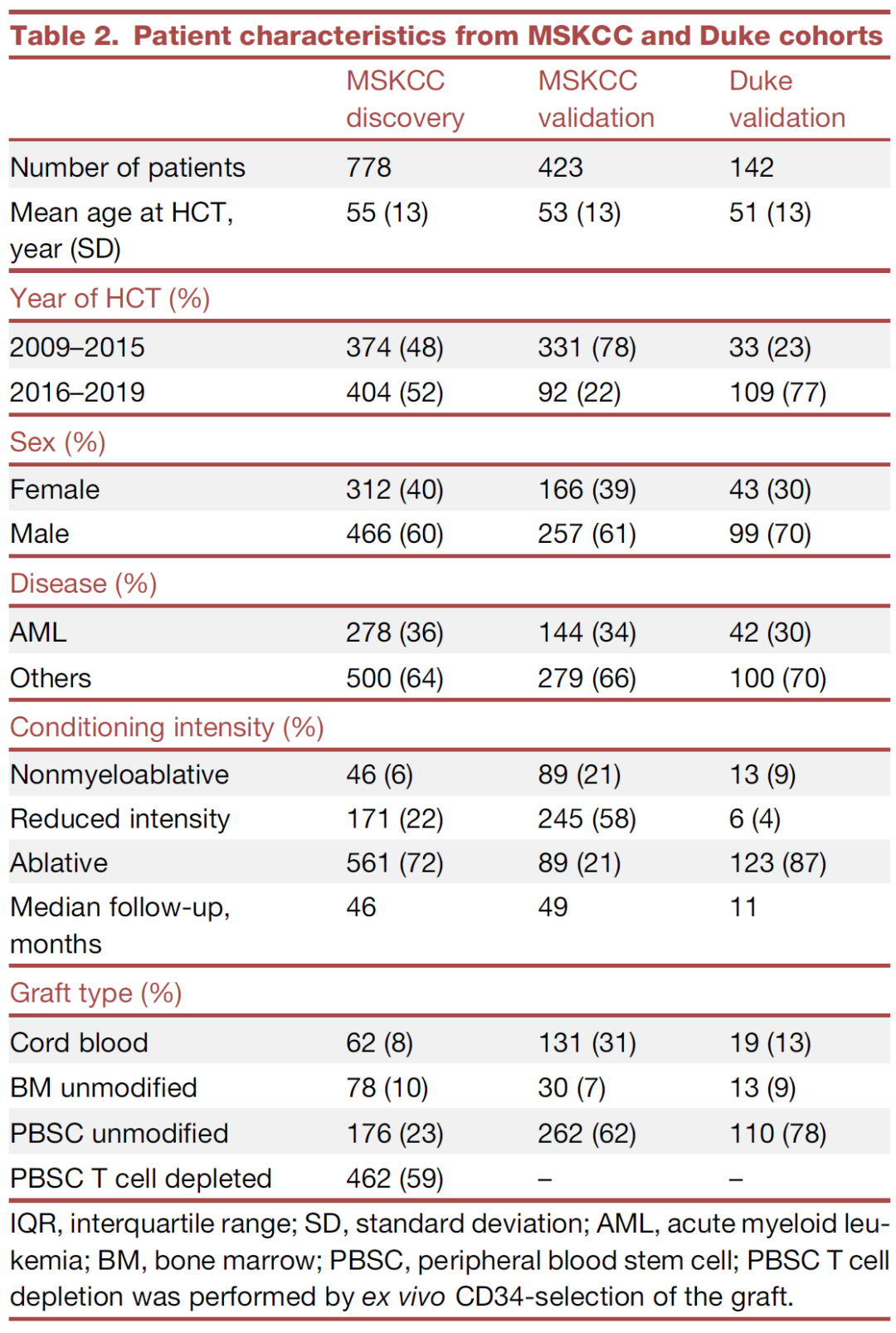
表2. 来自于MSKCC和杜克大学队列患者的特征描述
我们观察到基于药物暴露的患者特定的微生物响应分数在造血干细胞异体移植前14天和移植后14天之间与观察到的微生物类群相对丰富度和α多样性呈现显著正相关关系,这里的微生物类群相对丰富度和α多样性数据来自于MSKCC和杜克大学的验证组样本,样本收集时间段是在造血干细胞异体移植后第14天和第45天之间(图5C)。此外,在MSKCC和杜克大学的验证组造血干细胞异体移植患者样本中,预测具有较高肠球菌属(Enterococcus)相对丰富度增长的药物暴露患者,其全因死亡和移植相关的死亡风险也升高。特别的,在MSKCC验证组中,患者特定的肠球菌属(Enterococcus)响应分数也和GVHD相关的死亡风险升高呈显著正相关关系(图5D)。相反的,在MSKCC的发现队列和验证组造血干细胞移植患者样本中, 对于预测丹毒梭菌属(Eryispelatoclostridium)、布劳特氏菌属(Blautia)或α多样性维持相对稳定的药物暴露患者,其全因死亡在MSKCC的发现队列和验证组中的风险降低,同时在MSKCC的验证组中的GVHD相关的死亡风险也会降低(图5D)。总之,我们发现药物和微生物组的相关性对于造血干细胞异体移植之后未来微生物组的变化轨迹和临床治疗结果具有预测性。
此研究的分析框架是基于药物暴露会对肠道微生物产生影响,同时微生物会反过来改变临床结果这样一个假设。然而,在一些情况下,患者在不良结果风险较高时(与微生物组无关的复杂原因)可能会摄入影响肠道微生物的药物。为了进一步探究这种可能性,我们以肠球菌属(Enterococcus)为研究对象,在Cox比例风险模型中比较了肠球菌属(Enterococcus)相对丰度的风险比与相应的患者特定的响应得分。我们发现肠球菌属(Enterococcus)丰富度和死亡风险之间具有强相关性,这种相关性要强于患者特定的肠球菌属(Enterococcus)响应得分和整体死亡之间的相关性,特别是在MSKCC验证组中。然而,我们也观察到在控制肠道微生物组成的情况下患者特定的响应得分在预测死亡风险中仍然是一个统计上显著的预测因子。总之,这些结果表明药物暴露和临床结果之间的相关性部分独立于药物和肠道微生物之间的相互作用。
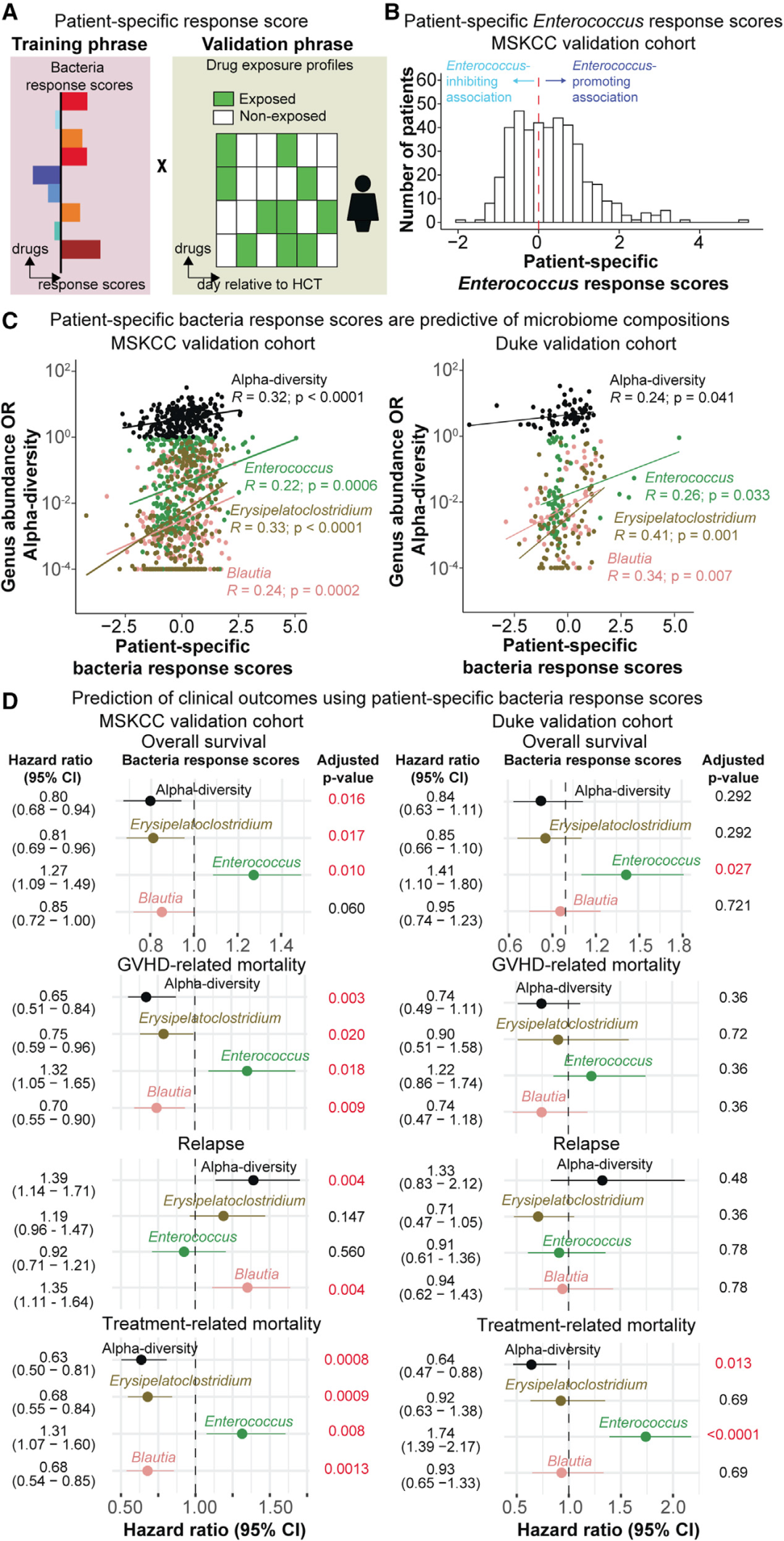
图5. 药物暴露的情况在两个独立的验证数据组中能够预测未来微生物组的变化轨迹和造血干细胞异体移植患者的临床结果
(A):患者特定的微生物响应得分计算示意图。
(B):验证组患者特定的肠球菌属(Enterococcus)响应得分,这一得分是仅仅根据药物暴露情况(造血干细胞异体移植前14天和移植后14天)和图3C中展示的微生物响应得分得到的。负分代表药物暴露情况和肠球菌属(Enterococcus)抑制效应有关,而正分代表药物暴露情况和肠球菌属(Enterococcus)促进效应有关。
(C):患者特定的微生物响应得分和观测得到的属水平相对丰富度或α多样性之间的皮尔森相关性系数。数据来自于MSKCC和杜克大学的验证组样本,样本收集时间段是在造血干细胞异体移植后第14天和第45天之间。
(D):在MSKCC和杜克大学的验证组造血干细胞异体移植患者样本中,患者特定的微生物响应得分能够预测全因死亡和移植相关的死亡风险,展示了Cox比例风险模型或Fine-Gray模型的结果,控制变量为年龄、性别、调理强度、移植来源和潜在疾病等。通过Benjamini-Hochberg矫正来调整p值。误差棒代表95%置信区间。
- 讨论 -
我们开发了PARADIGM计算方法来研究药物暴露和人类肠道微生物动态变化之间的相关性。这一方法的核心是分析了连续的肠道微生物组成状态是如何对抗生素和非抗生素类药物产生响应的。由于其它已经开发的计算方法主要研究的是抗生素类药物,我们的方法能够通过分析系统性收集的造血干细胞异体移植患者的大量样本进一步揭示许多非抗生素类药物和高通量测序得到的微生物动态变化之间的相关性。
我们的方法能够推断出药物暴露和微生物组之间有意义的相关性,尽管有许多混淆因子比如临床症状会促使药物的暴露,但是这对于选择使用药物去治疗肠毒性尤其重要。我们通过对比基于真实收集数据的微生物响应得分和模型模拟的结果来对我们的发现进行验证,结果表明我们的预测值和已经发表的数据结果具有强相关性。此外,只根据患者药物暴露情况计算得到的微生物响应得分能够让我们在两个独立的验证样本组中预测未来微生物的变化和患者的临床结果。这些结果显示PARADIGM方法能够得出与生物学和临床相关的假设。
综上,我们的研究为药理学的暴露和肠道微生物组成之间的相关性提供了进一步的见解。我们开发的PARADIGM算法能够鉴定出在生物学上具有意义以及在临床上相关的药物暴露和肠道微生物动态变化之间的相关性。PARADIGM方法有助于整合来自于多种类别的外源药物暴露、微生物动态变化和临床结果,以理解肠道微生物组健康的决定因素。这一计算框架适合于纵向收集的数据并且能够用于在未来进一步研究其它影响因子的作用,比如饮食或者其它相关的影响因子,或者应用到其它药物和微生物相互作用对临床结果非常重要的疾病研究中。
研究的局限性
本研究利用高通量测序的方法鉴定药物和微生物组之间的相关性,并且展现出利用这些相关性预测临床结果的能力。但是,本研究仍然存在一些局限性,在解读我们的结果时需要保持谨慎。局限性主要包括以下几个方面:(1) 本研究是一个回顾式的研究,需要开展控制实验去验证我们的预测是否具有因果关系。(2) 药物和微生物组的相互作用可能是独立于药物剂量的,这一点在本文中没有考虑。(3) 尽管已经有研究报道了药物协同和对抗的相互作用,但我们的模型假设每一种药物都是独立起作用的。(4) 移植的独特作用比如调理强度和移植方式在我们的模型中没有被完全考虑进去,我们假设这些效应可以被时间参数和药物治疗方案(取决于移植类型和调理方案)减弱一部分。(5) 药物暴露只是患者受到的各种干预措施中的一个组成部分,其它环境因子也会和肠道微生物组的变化相关,例如在健康个体和癌症患者中饮食都在改变肠道微生物中发挥着主要作用。未来的研究可以收集患者饮食摄入的数据来进一步阐明其与饮食之间的相关性。最后,我们认为PARADIGM方法会是一个研究微生物在有机体内动态变化的有用的工具,在未来可以应用到其它数据集中,包括药物暴露和其它环境因子数据,去复现或者进一步扩展在此研究中得到的研究结果。
参考文献
Nguyen C L, Markey K A, Miltiadous O, et al. High-resolution analyses of associations between medications, microbiome, and mortality in cancer patients[J]. Cell, 2023, 186(12): 2705-2718. e17.
- 作者简介 -
第一作者

纪念斯隆·凯特琳癌症中心
Chi L. Nguyen
Chi L. Nguyen,来自于纪念斯隆·凯特琳癌症中心,
来源链接:https://www.mskcc.org/research-areas/labs/members/chi-nguyen
通讯作者

纪念斯隆·凯特琳癌症中心
Marcel R.M. van den Brink
主任
Marcel R.M. van den Brink, 在莱顿大学获得医学博士学位,骨髓移植研究科学家,纪念斯隆·凯特琳癌症中心血液恶性肿瘤科主任,Alan N. Houghton主席。曾获得霍华德休斯医学院内科科学家奖(1996)、美国国家骨髓捐赠计划Amy Strelzer Manasevit学者奖(1999)、癌症研究基金达蒙·鲁尼恩学者奖(2001)、癌症研究基金会奖学金、入选美国临床研究学会(2004)、当选为美国医师协会会员(2013)、欧洲免疫学会联合会Immunology Letters Lecture Award (2014)、Delete Blood Cancer Award, DKMS (2015)、干细胞网络蒂尔和麦卡洛克讲座奖(2015)、莱顿大学Jaap de Graeff奖(2015)、荷兰皇家艺术与科学院外籍院士(2020),专门研究骨髓移植作为治疗血癌(如白血病)患者的方法。
来源链接:https://www.mskcc.org/cancer-care/doctors/marcel-van-den-brink#about_specialties
猜你喜欢
iMeta简介 高引文章 高颜值绘图imageGP 网络分析iNAP
iMeta网页工具 代谢组MetOrigin 美吉云乳酸化预测DeepKla
iMeta综述 肠菌菌群 植物菌群 口腔菌群 蛋白质结构预测
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature
一文读懂:宏基因组 寄生虫益处 进化树 必备技能:提问 搜索 Endnote
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流快速解决科研困难,我们建立了“宏基因组”讨论群,己有国内外6000+ 科研人员加入。请添加主编微信meta-genomics带你入群,务必备注“姓名-单位-研究方向-职称/年级”。高级职称请注明身份,另有海内外微生物PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。
点击阅读原文,跳转最新文章目录阅读


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









