






 2023年10月,厦门大学游伟伟团队开发了TOmicsVis R包,为转录组项目提供全面分析和可视化方案。该包基于46个R包设计40个函数,涵盖样本统计等6部分。还提供Shinyapp本地运行和在线服务,方便无编程经验者,满足常规转录组学项目分析需求。
2023年10月,厦门大学游伟伟团队开发了TOmicsVis R包,为转录组项目提供全面分析和可视化方案。该包基于46个R包设计40个函数,涵盖样本统计等6部分。还提供Shinyapp本地运行和在线服务,方便无编程经验者,满足常规转录组学项目分析需求。
点击蓝字 关注我们
TOmicsVis:具备Shinyapp界面的一站式转录组分析和可视化R包

iMeta主页:http://www.imeta.science
综 述
● 原文链接DOI: https://doi.org/10.1002/imt2.137
● 2023年10月13日,厦门大学游伟伟团队在 iMeta 在线发表了题为 “TOmicsVis: an all-in-one transcriptomic analysis and visualization R package with Shinyapp interface” 的文章。
● 本研究系统开发了TOmicsVis (Transcriptomics Visualization)R包(CRAN:https://cran.r-project.org/package=TOmicsVis,v2.0.0),为转录组项目提供全面的分析和可视化解决方案。
● 第一作者:苗奔奔、董炜
● 通讯作者:游伟伟(wwyou@xmu.edu.cn)
● 合作作者:韩兆方、骆轩、柯才焕
● 主要单位:厦门大学海洋与地球学院近海海洋环境科学国家重点实验室、中山大学附属口腔医院光华口腔医学院广东省口腔医学重点实验室、厦门大学福建海洋可持续发展研究院
亮 点
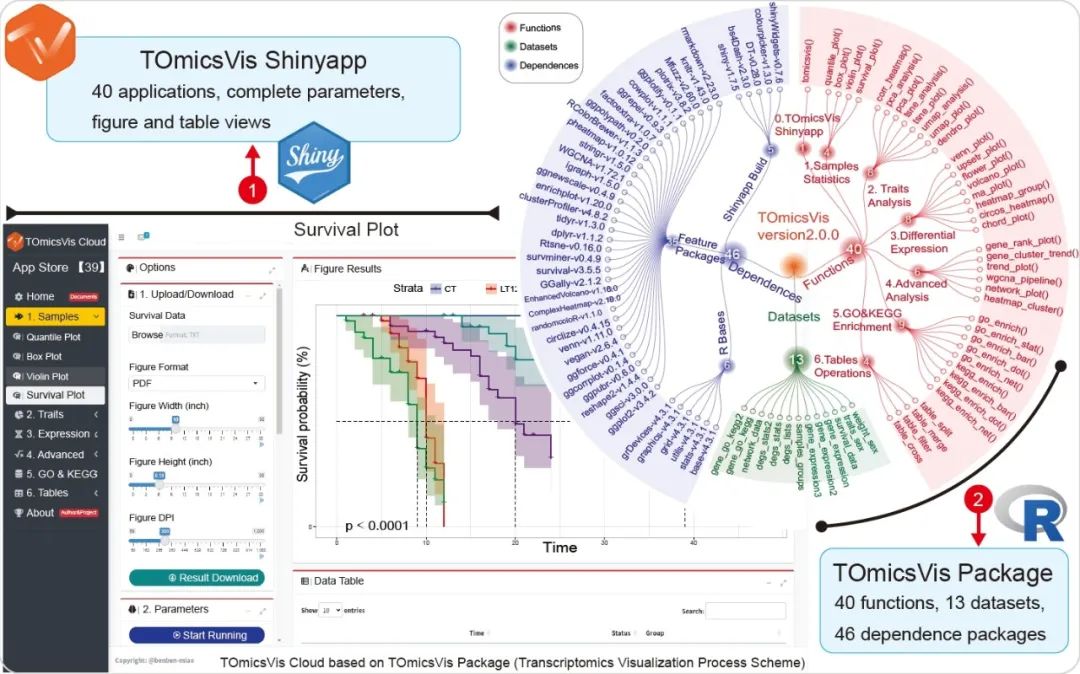
图 1. 图形摘要
● TOmicsVis R包为多分组转录组项目提供一站式分析和可视化流程,内置多分组示例数据集作为演示;
● TOmicsVis提供Shinyapp本地运行函数和在线界面分析服务,为无编程经验的科研者提供了随时交互分析的便利;
● TOmicsVis集成了基因表达趋势分析、基因聚类分析和加权基因共表达网络分析等多种高级分析和可视化方法。
摘 要
转录组学已被广泛应用于比较实验研究中,以揭示特定物种在不同实验条件下基因表达水平的生物学机制。然而,当前仍然缺乏一个精心设计的工具,用于优化和整合来自多个R包的分析和可视化功能。在本研究中,我们系统开发了TOmicsVis (Transcriptomics Visualization)R包(CRAN:https://cran.r-project.org/package=TOmicsVis,v2.0.0),为转录组项目提供全面的分析和可视化解决方案。它基于46个R包设计了40个精心打磨的函数,适用于多分组转录组项目的流程化分析,涵盖6个部分:样本统计(Sample Statistics)、性状分析(Traits Analysis)、差异表达(Differential Expression)、高级分析(Advanced Analysis)、GO和KEGG富集分析(GO and KEGG Enrichment)以及表格操作(Table Operation)。TOmicsVis Shinyapp可以在本地通过函数运行或在线(https://shiny.hiplot.cn/tomicsvis-shiny/)访问分析,为无编程经验的科研者提供了极大的便利。TOmicsVis提供了即用型的可视化函数和用户友好的交互界面,使科研者能够快速准确地分析和监控实验数据,以便于迅速深入探索转录组学数据。
视频解读
Bilibili:https://www.bilibili.com/video/BV1eG41127Mb/
Youtube:https://youtu.be/EdjPcJMqQdo
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
全文解读
引 言
在过去的十年中,转录组学已成为研究基因表达的一种标准而强有力的定量方法,在生物学研究中发挥着至关重要的作用。通过比较不同实验条件下的基因表达类型和丰度,研究人员可以筛选出与特定性状密切相关的差异表达基因(DEGs)。转录组学被广泛应用于研究生物不同生长阶段的基因表达、生物或非生物环境因素对生物体基因表达的影响。此外,非模式物种的转录组学研究可以不依赖于参考基因组,从而使得更广泛物种的基因表达谱和生物学特征得到探索和研究。由Illumina引领的下一代测序(NGS)技术和由PacBio为首的第三代测序(TGS)技术加速了转录组学的普及。测序技术的发展推动了基因组的破译和转录组学分析的迭代改进,进而促使了生物信息学分析工具的快速创新和更新。
转录组测序生成包含质量信息的FastQ文件,其需要一系列步骤进行转录本定量和下游分析。通常地,包括使用fastp程序进行序列过滤和HISAT2程序进行基因组比对。对于没有参考基因组的转录组分析,推荐使用Trinity程序进行转录本组装。在对所有样本的基因表达数据进行标准化后(使用FPKM或TPM等方法),使用DESeq2 R包来鉴定两两分组间的差异表达基因。韦恩Venn图和火山Volcano图适用于可视化差异表达基因的数量统计和差异分布。基因本体论(GO)和京都基因和基因组百科全书(KEGG)注释和富集分析对于探索基因功能至关重要,clusterProfiler R包提供基于超几何分布算法实现富集分析的流行方法。加权基因共表达网络分析(WGCNA)适用于基于所有转录本构建基因共表达模块,用于筛选与所研究性状密切相关的基因群。由于有许多R包可以实现转录组学不同步骤的分析和可视化功能,因此迫切地需要对其进行全面的整合和优化。
TOmicsVis旨在为转录组学提供全面的分析和可视化解决方案,捕获数据分析过程中生物信息在数据间的传递和转换,优化和集成一系列分析和可视化功能,从而产出预发表的表格和图像结果。在转录组下游分析中,除差异表达分析之外,还包含各种高级分析方法用于数据挖掘。TOmicsVis为每个步骤提供备选方案,如在数据降维分析中提供多种算法:主成分分析(PCA)、t-分布随机邻近嵌入(t-SNE)和统一流形逼近与投影(UMAP),可分析生成降维结果表格和图像。TOmicsVis Shinyapp提供了完整的用户操作界面,可辅助科研者快速高效地进行转录组数据分析和可视化工作。
结果与讨论
分类概览和可视化函数
TOmicsVis共依赖46个R包进行开发,其中包括6个base包,5个用于构建Shinyapp的包,其余35个第三方包用于实现数据处理、转录组分析和定制可视化。基于这些依赖,TOmicsVis精心开发了40个适用于转录组项目流程化数据分析和可视化的函数,涵盖从样本性状到基因表达分析。这些分类包括:样本统计(Sample Statistics)、性状分析(Traits Analysis)、差异表达(Differential Expression)、高级分析(Advanced Analysis)、GO和KEGG富集分析(GO and KEGG Enrichment)和表格操作(Table Operation)。一个特殊的函数tomicsvis专为TOmicsVis R包定制,用于启动本地化的Shinyapp用户界面。基于已发表的多分组转录组学研究文章,提供了13个内置于TOmicsVis包中的示例数据集。这些数据集被用于函数示例测试,方便用户快速准备数据和预览函数功能(图2A)。
样本统计分类中的功能用于执行统计和检测可测量性状之间的差异,并对环境胁迫生存记录数据进行生存分析。在性状分析分类中,包括基于Pearson或Spearman算法的相关性分析,以及基于PCA、t-SNE、和UMAP算法的降维分析和Ward分层聚类分析。在差异表达分类中,韦恩Venn、UpSet、热图Heatmap、圈状热图和弦图Chord用于可视化多分组间的差异表达基因。火山图Volcano和M-vs-A(MA)图适用于展示两个配对分组间的差异表达基因分布。高级转录组分析面向所有基因,并将深度数据挖掘扩展到差异表达分析之外。例如,gene_cluster_trend函数首先对所有组中的所有基因进行聚类分析,然后构建表达趋势一致的多个基因表达图谱。wgcna_pipeline函数对所有基因执行聚类分析后,基于WGCNA v1.72.1包构建与所研究的性状密切相关的基因共表达模块。GO和KEGG 富集分析分类中的函数基于clsterProfiler v4.8.2包进行改写。基于参考和非参考基因组基因注释结果对GO和KEGG注释表进行规范化,将数据处理和富集分析构建生成新的函数。
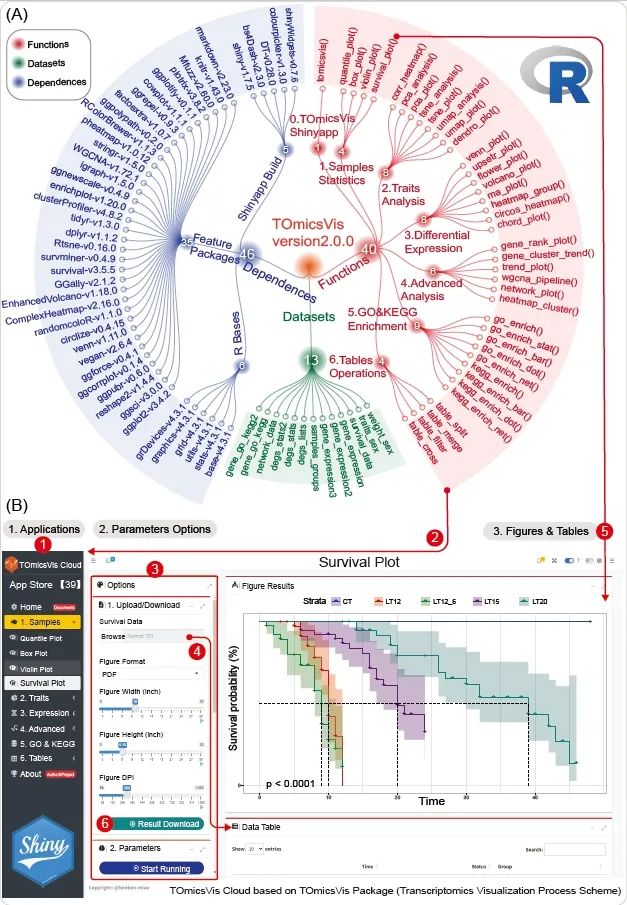
图 2. TOmicsVis R包和TOmicsVis Shinyapp的逻辑架构
(A)TOmicsVis基于46个R包开发转录组数据分析及可视化的40个函数,及内置13个多分组转录组示例数据集。可视化函数用红色高亮显示,依赖包用蓝色高亮显示,内置示例数据集用绿色高亮显示;(B)TOmicsVis Shinyapp用户界面。编号1-6表示Shinyapp执行步骤,1.启动侧边栏应用程序;2.调用TOmicsVis R包函数;3.切换到参数面板修改参数;4.用户数据上传输入;5. 示例中演示执行survival_plot函数;6.下载可视化PDF结果文件。
TOmicsVis Shinyapp的实用性
为使得TOmicsVis R包功能对无编程经验的科研者更加人性化,我们基于擅长Web界面设计和运行的R包shiny v1.7.5(https://github.com/rstudio/shiny/)、bs4Dash v2.3.0(https://github.com/RinteRface/bs4Dash/)等开发了一个友好的交互式分析操作界面平台。安装完TOmicsVis包后,用户可以使用tomicsvis函数在本地启动和部署Shinyapp,此时所有计算都将调用本地计算机资源,包括内存、CPU和存储。更便捷的是,我们还提供了免费的在线分析服务和计算资源,供科研者可随时随地访问和执行分析任务(https://shiny.hiplot.cn/tomicsvis-shiny/)。
TOmicsVis Shinyapp用户界面从左到右依次为应用菜单侧边栏、参数调节面板、图像和表格显示面板(图2B)。应用菜单栏的Home和About页面分别显示TOmicsVis API文档和项目信息,其余6个折叠菜单包含对应的所有功能的应用程序。TOmicsVis Shinyapp用18000+行R代码编写,能够实现所有40个函数对应的完整分析和可视化功能。通过点击侧边菜单栏启动应用程序后,参数操作面板、图像和表格显示面板会相应地切换。参数操作面板从上到下分为数据上传和下载参数、分析和可视化参数两个部分。得益于Shiny框架提供的各种文件上传下载、文本、数值、选项、颜色和按钮Web组件,所有函数的参数皆提供了特定的交互组件。最终结果可以在图像显示面板中查看,图像结果可以下载为PDF或JPEG格式(默认DPI值为300),表格结果仅支持下载为TXT格式。综上所述,TOmicsVis Shinyapp的分析和可视化功能得益于TOmicsVis R包开箱即用的函数和示例数据,两者相辅相成,有利于长期维护和深入开发工作。
TOmicsVis资源和竞争力
TOmicsVis采用通用公共许可证(GPL)许可开源项目,R包、API文档、网站和Shinyapp的所有源代码都存储在GitHub存储库https://github.com/benben-miao/TOmicsVis/。TOmicsVis通过综合R存档网络(CRAN)平台(https://cran.r-project.org/package=TOmicsVis)提供并维护着稳定版本,最新功能的不稳定版本可在GitHub存储库上获得。此外,我们还创建了综合API文档和教程,包括文本和视频内容(https://benben-miao.github.io/TOmicsVis/)。TOmicsVis Shinyapp(https://shiny.hiplot.cn/tomicsvis-shiny/)与R包协同开发并同步长期维护,为用户提供稳定的在线分析服务。正因如此,自发布以来一个月的时间里,TOmicsVis已经获得超过700次唯一安装的显著吸引力(截至2023-09-13)。
为了全面地综述TOmicsVis的优势,我们将其与其它几个常用的转录组分析相关R包和Web服务进行了比较(表1)。R包systemPipeR v2.6.3、RNASeqR v1.0.3和bcbioRNASeq v0.5.5的优势在于,它们包括基于原始测序数据Reads的转录组分析的全流程。而不同地,TOmicsVis侧重于下游数据分析和极致的可视化,适用于多分组转录组项目。TOmicsVis包括基于算法的基因表达聚类和趋势分析方法,除差分表达分析之外,还提供了多种候选基因深度挖掘和可视化方法。与Hiplot和ImageGP生物信息学Web服务相比,TOmicsVis Shinyapp也提供用户友好的交互式Web界面。TOmicsVis的独特之处在于,作为一个开源的R包,它可以不断接受更多的优化,并方便被其它R包或脚本调用,以增强其对转录组分析中的作用。总之,TOmicsVis的目标是创建一个优化的适用于多分组转录组项目下游分析R包,并基于这些函数提供本地和在线的用户界面分析平台。
表1. TOmicsVis与5个转录组学功能相关的R包或网站的功能比较

TOmicsVis用户案例
案例1:实验中的样本统计和性状分析
为了演示TOmicsVis在多分组转录组学研究项目中的有效性和优势,我们基于已发表的研究文章中的数据展开三部分案例分析。
在完成野外或实验室的采样后,我们使用基于95%可信区间的分位数图quantile_plot函数对所有200个样本的体重进行正态分布检验。该函数允许用户控制多面板的整合或分离,从而进行分组展示(图3A)。随后,我们对多个可测量性状进行统计分析和显著性差异检测。此时箱线图box_plot和小提琴图violin_plot都是可行的选择,它们支持在结果图像中注释比较组间的显著性p值。结果表明配对性状之间存在显著的统计学差异(p < 0.05)(图3B,C)。如果涉及极端环境应激或毒性测试的实验,我们可以使用生存分析survival_plot函数有效地分析和解释样本存活状态随时间的记录数据。结果表明胁迫温度越低存活率越低,而正常室温下样本存活率不受影响(图3D)。综上所述,TOmicsVis强调了实验过程中可测量性状数据的准备,并在结果的分析和可视化中充分考虑了数据的复杂性和科研者的实际需求。
我们使用相关性热图corr_heatmap函数对所有基因表达数据执行Pearson、Spearman或Kendall相关性分析,获得组内和组间所有样本之间的相关性,并将其以热图的形式展示。其独特性在于中位数通常对应于颜色调过渡颜色值,允许用户显示只有正值或负值的相关性系数。分析结果显示,组内样本表现出比组间样本更高的相关性(图3E)。降维算法用于基于大量基因揭示样本之间的分布关系,本案例中的基因表达矩阵来自5个组的15个样本包含11,033个基因。PCA降维算法被广泛使用,TOmicsVis提供两个函数pca_analysis和pca_plot分别用于PCA分析并输出分析结果表格和图像。该功能允许用户指定任何两个PC值用于展示占比及贡献度,并为每个组添加代表95%置信区间的椭圆形背景(图3F)。此外,t-SNE和UMAP降维算法被推荐用于单细胞分析流程,因此这两种算法的分析和可视化功能包含在TOmicsVis中(图3G)。聚类算法对于分类样本仍然有效,dendro_plot函数首先执行聚类分析,并显示样本之间距离关系的层次进化树(矩形、圈状或系统发育轨迹)。它默认使用欧几里得Euclidean或堪培拉Canberra距离方法,以及沃德Ward's的层次聚集聚类算法。结果显示样本聚类层级树与实验采样方案高度一致(图3H)。综上所述,TOmicsVis集成了Pearson's、PCA、t-SNE、UMAP、Euclide和Ward's分层聚集聚类等多种算法,从相关性、降维和聚类的角度探索分组和性状特征。
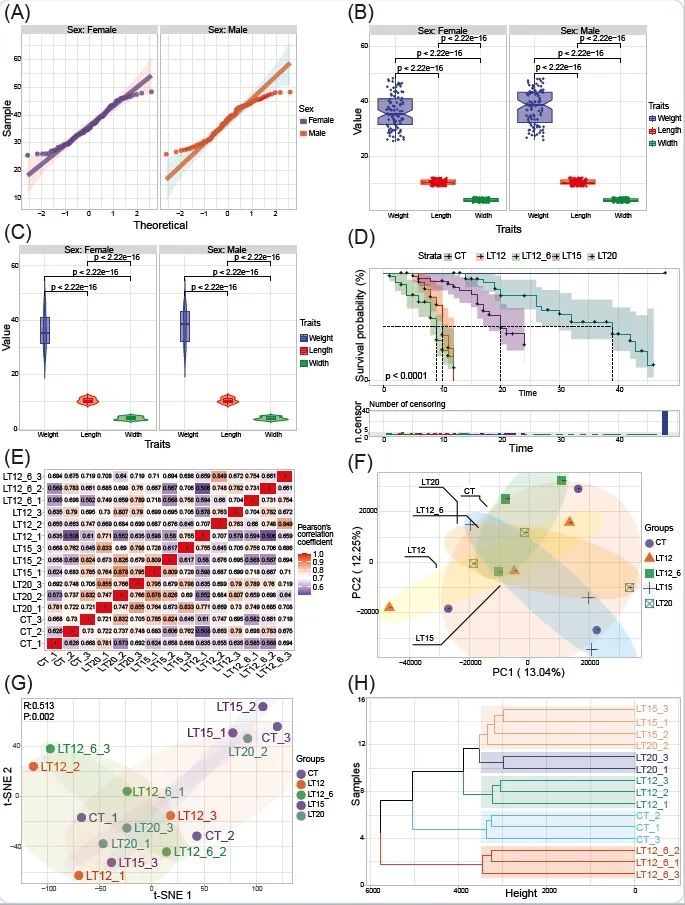
图3. TOmicsVis实现的样本统计和性状分析
(A)分位数图;(B)箱线图;(C)小提琴图;(D)生存分析图;(E)相关性热图;(F)PCA分析图,图上显示95%的置信区间;(G)t-SNE分析图,p值和R值显示在图左上角;(H)分层聚类树状图。上述结果是使用TOmicsVis分析包含5组真实转录组项目数据获得,这些数据也被用作内置数据集。可以通过修改函数的参数来定制图像中的元素和颜色等细节。
案例2:多分组差异表达分析筛选候选基因
在转录组学研究中,差异表达基因的鉴定和功能解析是分析过程的主要焦点。差异表达基因的生物学功能和参与的生物学过程可以帮助科研者理解特定实验条件对基因转录水平和生物体内生物学过程的影响。
基于5个分组的转录组数据,将CT组和其他四个低温胁迫实验组之间进行差异表达分析。在这四个成对比较中鉴定差异表达基因,使用venn_plot函数可视化这些所有比较中唯一和共享的差异表达基因。该函数适用于最多7组数据交集分析。结果表明CT. vs.LT12_6比较差异表达基因数量最多,表明较低的温度和较长的胁迫时间引起更多的基因发生差异表达。在所有四个比较中共有306个差异表达基因共享(图4A)。鉴于处理更多分组时需要数据交集分析,upsetr_plot函数可以弥补venn_plot的局限性(图4B)。在所有比较对差异表达分析的结果统计,volcano_plot函数使用|Log2FoldChange| > 1和p ≤ 0.05(即:-Log10Pvalue ≥ 1.30103)的默认条件进行差异表达基因筛选。符合差异表达筛选条件的基因由红点表示,而蓝色、绿色和灰色点分别表示仅满足p值、Log2FoldChange或不满足过滤条件的基因。结果中部分重要的差异表达基因用基因名称标记,以方便科研者关注和分析(图3C)。MA可视化由ma_plot函数执行,其在鉴定和注释差异表达基因的目的角度与volcano_plot相似(图4D)。
基于筛选的306个共享差异表达基因,我们展开进一步分析以探索所有样本的基因表达差异。实际上,作为演示我们只选择了前30个基因。函数heatmap_group最初对所有15个样本进行聚类分析,并分别以聚类树和热图的形式呈现样本聚类和基因表达的结果。该函数允许用户调整坐标轴标题的方向、热图颜色等细节元素(图4E)。当需要显示更多基因时,矩形热图受到空间的限制,因此圈状热图circos_heatmap函数可以作为heatmap_group的补充可视化方法。显然在图4F所示的结果中,圈状热图中显示50个差异表达基因,这些基因名称仍然清晰可见。将基因表达值显示为chord_plot中弦线的宽度,有效地显示了30个差异表达基因在所有15个样本中的表达特征。因此,差异表达基因CDO1、APOC2、ACAN和ACAA2值得关注(图4G)。经过一系列差异表达分析,表达变化最高的基因不容忽视,gene_rank_plot函数根据Log2FoldChange对所有差异表达基因进行排名,并筛选出上调和下调倍数变化最高的Top-10基因进行显示(该值是可调的)。YME1L1和JWE等差异表达基因值得进一步验证(图4H)。
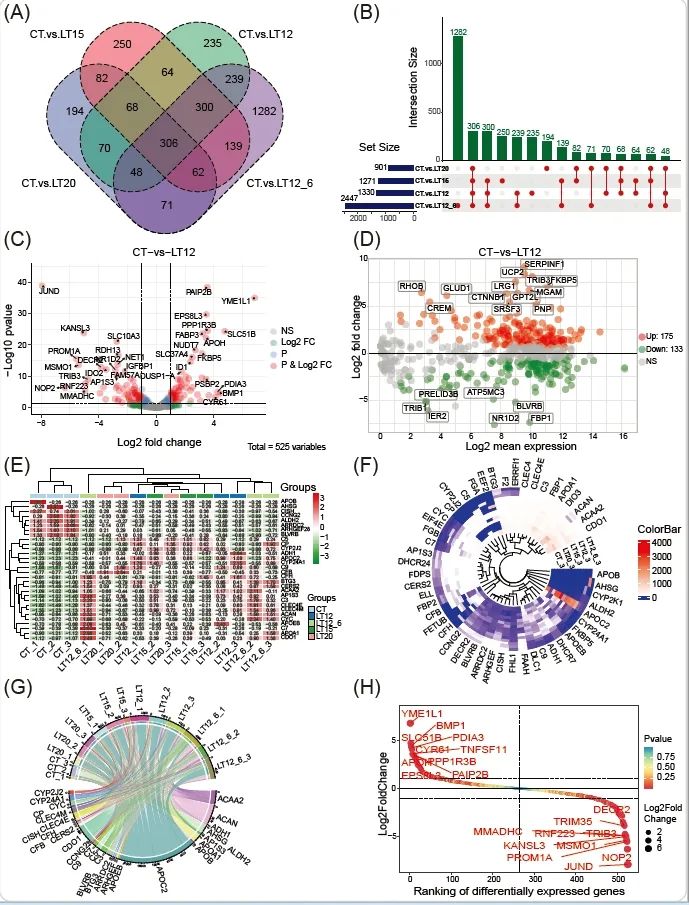
图4. 组间基因的差异表达分析和可视化
(A)韦恩Venn图;(B)UpSet图;(C)火山Volcano图;(D)MA散点图;(E)多分组热图;(F)圈状热图;(G)弦Chord图;(H)基因排序图。上述结果是使用TOmicsVis分析包含5组真实转录组项目数据获得,这些数据也被用作内置数据集。可以通过修改函数的参数来定制图像中的元素和颜色等细节。
案例3:通过构建共表达模块和趋势挖掘基因
转录组学高级分析旨在发现更多潜在的基因表达模式,特别是在涉及多个分组(≥ 3)的研究中。目前有两种主要的方法正在探索基因表达趋势,通过聚类分析构建基因簇,将具有相似表达特征的基因聚类,这提供了潜在的生物学见解。
函数gene_cluster_trend实现了基于Mfuzz v2.60.0 R包提供的一系列功能的全面分析和可视化功能,如基于基因表达矩阵创建表达集合、数据归一化、执行聚类算法和可视化趋势图。结果中我们构建了四个基因表达趋势一致的基因簇,每个簇中的基因在所有组中显示出相似的表达趋势,Cluster 1和Cluster 3中的基因上调,Cluster 2和Cluster 4中的基因下调(图5A)。函数trend_plot不执行数据聚类分析,而是将数据归一化,以探索组间的基因表达趋势。归一化方法包括std(标准)、globalminmax(全局最小和最大值)、centerObs(中心观测值)等,它还支持以KEGG途径作为分类(图5B)。
我们基于WGCNA官方文档创建了一个加权基因共表达网络分析管道,包括数据准备和预处理,筛选构建共表达模块的最佳软阈值β,分析模块之间的皮尔逊Pearson相关性,以及模块和性状之间的皮尔逊Pearson相关性热图。最终,函数运行生成多个关键的表格和图像结果(图像结果提供PDF和JPEG格式)。在模块-性状相关热图结果中,MEmidnight Blue模块(R=0.79,p=5e-4)和MEgrey模块(R=0.7,p=0.004)中的基因与LT12组(低温性状)高度相关。MEturquoise模块(R=0.9,p=5e-6)和MEbran模块(R=0.8,p=3e-4)中的基因与LT12_6组(低温性状)高度相关(图5C)。利用network_plot函数构建了WGCNA分析获得的前200个基因对之间的相互作用网络,与Cytoscape v3.10.1程序相比,其基于inbound和outbound连接度实现网络构建,并提供多种网络结构。结果默认为球体结构展示,其中CYR61、CEBPD和PPP1R3G都属于中心枢纽基因,与其它基因密切互相作用(图5D)。函数heatmap_cluster结合聚类和趋势分析,允许用户指定聚类数量。结果中创建了5个聚类,Cluster 1、Cluster 2和Cluster 3中的基因表达上调,Cluster 3的基因数量最高(图5E)。
基因注释和富集分析是获取基因生物学功能信息的主要方法。利用clusterProfiler R包进行基于超几何分布算法的富集分析是方便的,特别是对于来自模式物种的注释信息。在实际的转录组项目中,有两种主要的方法来获得GO和KEGG注释数据:a.来自未包含在模式物种注释数据库中的新的或私有参考基因组的注释信息。b.对于没有参考基因组的物种,转录本被组装后通过BLAST针对GO和KEGG数据库获得注释信息。最终,我们标准化了通过各种方法获得的GO和KEGG注释表,并重新设计了用于富集分析和可视化的函数。为研究人员提供用于富集分析的表格和图像结果。函数go_enrich_stat执行GO富集分析和统计可视化(图5F)。函数kegg_enrich_bar和kegg_enrich_net分别基于KEGG富集分析输出条形图和网络图(图5G,H)。
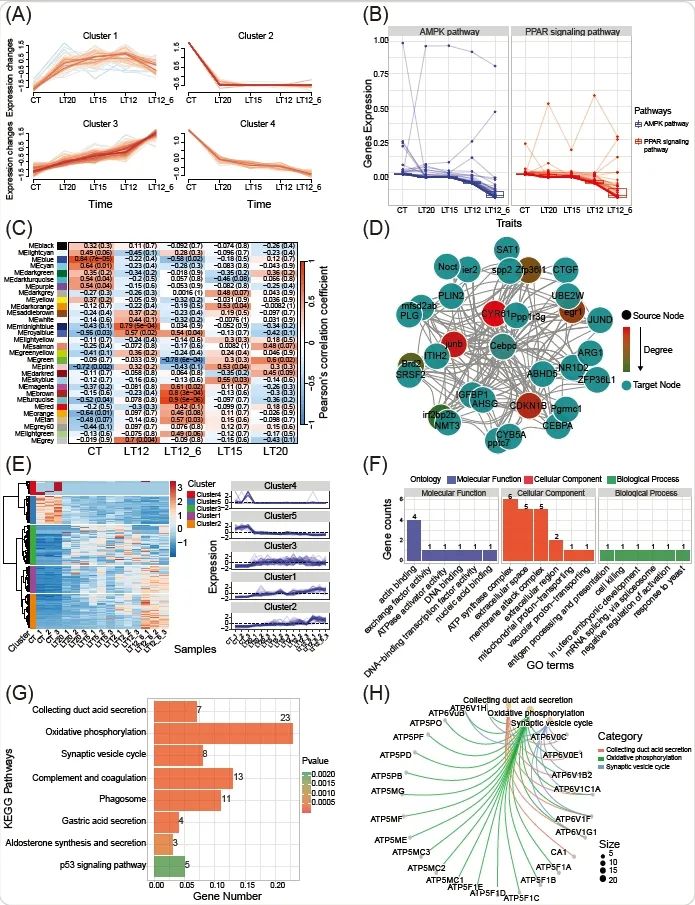
图5. 转录组高级分析
(A)基因聚类和趋势;(B)基因表达趋势;(C)共表达模块和性状相关性热图;(D)基因间的相互作用网络;(E)基因聚类热图和趋势整合;(F)GO富集结果统计图;(G)KEGG富集结果条形图;(H)KEGG富集网络图。上述结果是使用TOmicsVis分析包含5组真实转录组项目数据获得,这些数据也被用作内置数据集。可以通过修改函数的参数来定制图像中的元素和颜色等细节。
方 法
TOmicsVis用于流程演示的内置数据集
TOmicsVis内置的转录组示例数据集允许随时测试函数功能和参数细节。这些数据集是根据已发表的转录组学研究文章整理的,其中包括25℃室温(CT)、20℃、15℃、12℃和12℃低温应激6小时组(LT20、LT15、LT12、LT12_6),每组包含三个重复,共15个样本。本研究可以有效地测试TOmicsVis在多分组转录组学研究中的适用性和优势,仅包括两组的实验数据仍然适用。我们通过使用样本、性状、基因表达数据等根据实际情况进行标准化,因此用户可以在实验中快速准备数据(表2)。其中,weight_sex、traits_sex、survival_data包含样本测量性状的信息,gene_expression、gene_expression2、gene_expression3和samples_groups包含所有样本的基因表达数据。degs_lists、degs_stats、degs_stats2包含差异表达基因的统计数据,gene_go_kegg、gene_go_kegg2包含基因的GO和KEGG注释信息,network_data包含基因的相互作用数据。这些数据集将伴随TOmicsVis R包一起安装,且这些表格数据可以从GitHub源代码仓库获得(https://github.com/benben-miao/TOmicsVis/tree/main/inst/data-tables/)。
表2. TOmicsVis包内置转录组数据集。所有数据集都是函数默认数据,可在 https://github.com/benben-miao/TOmicsVis/tree/main/inst/data-tables/获得
Data NameDescriptionFunctions

开发需要的基础设施
TOmicsVis基于R v4.3.1开发,利用roxygen2 v7.2.3包生成和更新API文档。函数的正确性和软件包的完整性是使用devtools v2.4.5包执行,基于DESCRIPTION和NAMESPACE文件执行编译。API文档和网站基于pkgdown v2.0.7和_pkgdown.yml配置文件构建,该文件提供在线帮助文档(https://benben-miao.github.io/TOmicsVis/)。对于数据操作,来自tidyverse生态系统的dplyr v1.1.2和tidyr v1.3.0包用于转换数据结构。TOmicsVis优先使用ggplot2 v3.4.2,这是一个广泛使用的绘图R包,提供可定制的可视化功能。此外,ggsci v3.0.0软件包用于提供常用的颜色调色板。
结 论
TOmicsVis R包使用46个R包设计了40个适用于多分组转录组项目分析及可视化功能,并提供13个包含5个分组实验的RNA-Seq示例数据集。这些功能分为6类:样本统计(Sample Statistics)、性状分析(Traits Analysis)、差异表达(Differential Expression)、高级分析(Advanced Analysis)、GO和KEGG富集分析(GO and KEGG Enrichment)和表格操作(Table Operation),有效地满足了常规转录组学项目分析的需求。TOmicsVis R包在CRAN(https://cran.r-project.org/package=TOmicsVis)上进行维护,累积700+次的唯一下载统计(截至2023-09-13)。此外,提供了Shinyapp的本地运行版本和在线分析服务(https://shiny.hiplot.cn/tomicsvis-shiny/),为无编程经验的科研者提供便利。
代码和数据可用性
TOmicsVis R包、Shinyapp的源代码和GitHub存储库上的示例数据集:https://github.com/benben-miao/TOmicsVis/,API文档和教程网站:https://benben-miao.github.io/TOmicsVis/,TOmicsVis 稳定版本在 CRAN平台发布:https://cran.r-project.org/package=TOmicsVis,在线Shinyapp 分析服务:https://shiny.hiplot.cn/tomicsvis-shiny/。补充材料(图形摘要、幻灯片、视频、中文翻译版本和更新材料)可在在线DOI或iMeta Science http://www.imeta.science/中找到。
引文格式:
Benben, Mao, Wei Dong, Zhaofang Han, Xuan Luo, Caihuan Ke and Weiwei You. 2023. “TOmicsVis: an all-in-one transcriptomic analysis and visualization R package with Shinyapp interface.” iMeta. e137. https://doi.org/10.1002/imt2.137
作者简介

苗奔奔(第一作者)
● 厦门大学海洋与地球学院海洋生物技术专业博士在读。
● 致力于海洋生物多组学研究和生物信息学开发。作为Hiplot生物信息分析云平台(https://hiplot.cn),OmicsSuite多组学桌面套件(https://github.com/omicssuite/),TOmicsVis转录组学可视化R包(https://github.com/benben-miao/TOmicsVis/)开发者,相关学术成果已发表于Briefings in Bioinformatics、Animals、Frontiers in Marine Science、iMeta等期刊。

董炜(第一作者)
● 中山大学附属口腔医院光华口腔医学院博士后。
● 主要致力于单细胞多组学、T细胞免疫记忆研究和生物信息学开发。先后参与Hiplot生物信息分析云平台(https://hiplot.cn),OmicsSuite多组学桌面套件(https://github.com/omicssuite/)和TOmicsVis转录组学可视化R包(https://github.com/benben-miao/TOmicsVis/)的开发,以第一或共同第一作者在Briefings in Bioinformatics, Genomics Proteomics & Bioinformatics, iMeta和Genome Biology and Evolution等学术期刊发表SCI论文7篇,另在Nature, Cell, Journal of Clinical Investigation和Plant Biotechnology Journal等国际权威期刊参与发表学术论文9篇,谷歌学术他引670余次,H-index 10。

游伟伟(通讯作者)
● 厦门大学海洋与地球学院教授,博士生导师。
● 致力于研究贝类基因组学与遗传育种,及海洋贝类对全球变化的响应。已在Reviews in Aquaculture、Molecular Ecology Resources、Aquaculture、Science of the Total Environment等期刊以第一作者、通讯作者或参与发表学术论文180余篇。
更多推荐
(▼ 点击跳转)
iMeta | 引用7000+,海普洛斯陈实富发布新版fastp,更快更好地处理FASTQ数据
iMeta | 德国国家肿瘤中心顾祖光发表复杂热图(ComplexHeatmap)可视化方法


1卷1期

1卷2期

1卷3期

1卷4期

2卷1期

2卷2期

2卷3期

2卷4期
期刊简介
“iMeta” 是由威立、肠菌分会和本领域数百位华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表原创研究、方法和综述以促进宏基因组学、微生物组和生物信息学发展。目标是发表前10%(IF > 15)的高影响力论文。期刊特色包括视频投稿、可重复分析、图片打磨、青年编委、前3年免出版费、50万用户的社交媒体宣传等。2022年2月正式创刊发行!
联系我们
iMeta主页:http://www.imeta.science
出版社:https://onlinelibrary.wiley.com/journal/2770596x
投稿:https://mc.manuscriptcentral.com/imeta
邮箱:office@imeta.science


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









