






点击蓝字 关注我们
干旱荒漠地衣结皮关键微生物组的宿主选择趋势

iMeta主页:http://www.imeta.science
研究论文
● 原文链接DOI: https://doi.org/10.1002/imt2.138
● 2023年10月10日,中国科学院微生物研究所魏鑫丽团队在 iMeta 在线发表了题为 “Host selection tendency of key microbiota in arid desert lichen crusts” 的文章。
● 本研究揭示了干旱荒漠地衣关键微生物组的组成规律、追溯了地衣相关细菌特别是关键物种的来源,为更好地了解地衣共生体的稳态维持机制提供了重要参考。
● 第一作者:张婷婷
● 通讯作者:魏鑫丽(weixl@im.ac.cn)
● 合作作者:Martin Grube
● 主要单位:中国科学院微生物研究所、中国科学院大学、奥地利格拉茨大学
亮 点
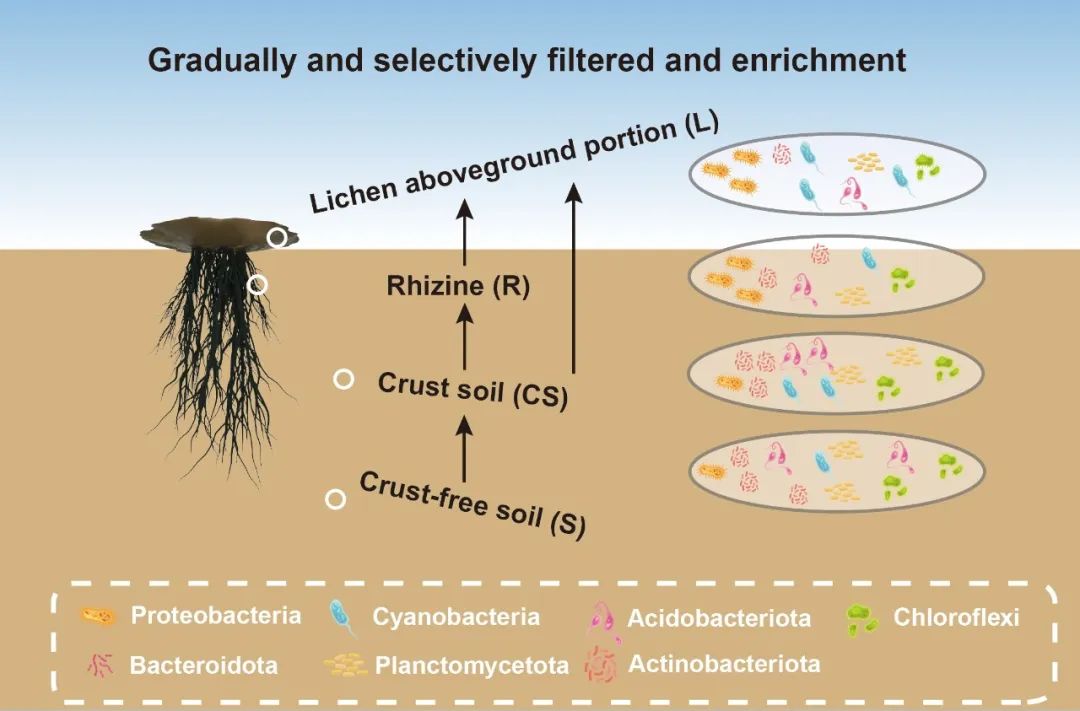
● 石果衣地衣结皮中的细菌群落组装主要受垂直取样部位的影响;
● 石果衣地衣体中的细菌主要来自结皮土,并逐级过滤;
● 石果衣地衣体中的关键细菌伯克氏菌目Burkholderiales,Pyrinomonadales,根瘤菌目Rhizobiales,鞘氨醇单胞菌目Sphingomonadales和土壤红杆菌目Solirubrobacterales具有宿主选择趋势。
摘 要
地衣作为生物土壤结皮的重要组成部分,在荒漠生态系统中起着不可忽视的固沙作用。然而,人们对这种被称为微型生态系统的地衣其群落组装及其与环境的关系知之甚少。本研究以石果衣属地衣Endocarpon为对象,利用16S rDNA基因V4高变区的扩增子测序,首次对来自三块代表荒漠的120个样品的4个垂直生态取样位(地衣地上部分L、地衣地下假根部分R、结皮土SC和流沙土S)中的细菌群落进行了分析。结果表明,石果衣结皮垂直尺度上的细菌群落组装主要受取样部位影响,其次是地点,而与地衣物种无关。变形菌门Proteobacteria是L部位中的优势细菌类群,与S部位相比,L和R部位富集根瘤菌目Rhizobiales中的拜叶林克氏菌科Beijerinckiaceae和黄杆菌科Xanthobacteriaceae。溯源分析表明,地衣中的细菌主要来源于结皮土,并被逐级选择性过滤。差异指数(DSI)值从CS到R,再到L部位,逐渐增大,L部位的DSI值最大,表明地衣宿主对细菌群落的过滤作用在取样部位中从下到上逐渐增加。微生物共存网络分析表明地衣体中细菌群落具有稳定的网络结构。本研究揭示了干旱荒漠地衣关键微生物组的组成规律、追溯了地衣相关细菌特别是关键物种的来源,为更好地了解地衣共生体的稳态维持机制提供了重要参考。
视频解读
Bilibili:https://www.bilibili.com/video/BV12B4y1R7ap/
Youtube:https://youtu.be/i7tvYWFoYaE
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
全文解读
引 言
土地荒漠化范围的急剧扩大是全世界最严重的问题之一。荒漠地区因为气候环境干旱、土壤养分贫乏,维管植物覆盖率较低,而生物土壤结皮(BSC)独具优势。BSC覆盖了70%的干旱和半干旱地区,约占地球陆地表面的40%,由蓝细菌、藻类、地衣、微型真菌、苔藓组成,其中地衣在促进营养循环和改善土壤的物理稳定性等方面发挥着极为关键的作用。
地衣是由真菌(共生菌)与藻类和/或蓝细菌(光合共生物)组成的共生体,同时还有共存着细菌和真菌等相关微生物。荧光原位杂交结果显示地衣体内细菌多样性高且存在非常普遍。宏基因组分析结果表明,地衣相关的细菌可能参与营养供应、抵抗生物和非生物胁迫、光合作用、解毒和地衣体降解等生命活动。简言之,地衣不仅与相关细菌密不可分,其本身还可能作为恶劣环境中保存细菌物种的“庇护所”。地衣相关的细菌群落结构可能是由多种因素塑造,如地衣附着的基物类型、地衣体年龄和生长的周围环境。然而,地衣相关细菌群落的组装机制是否与地衣共生体有关,以及地衣共生体如何维持其稳态尚不清楚。
一些研究已经调查了树生地衣和石生地衣中的细菌种类组成,但很少涉及其可能的来源,因此很难探究细菌组与地衣共生体的潜在关系。对土生地衣的研究主要集中在群落组成和形成的结皮理化性质方面,而且,这些研究中的地衣结皮都取自距离地表几毫米厚度内,取样包括了沙土并未考虑地衣形态上的细节,这可能会对分析结果产生偏差,因为样品中很可能混杂了附着于地衣体的假根或茸毛生上的结皮土相关微生物。尽管人们越来越关注生物土壤结皮中的地衣,但对土生地衣的微生物群落特征、共存网络结构和群落构建的潜在驱动因素缺乏足够的了解。
为了回答这些问题,我们选择了石果衣属Endocarpon地衣结皮进行测序分析,主要是因为它们在干旱、半干旱地区荒漠中分布广泛并具有显著的固碳和固沙能力。我们在中国荒漠中收集了两种石果衣结皮,分别是卷边石果衣Endocarpon adsurgens和石果衣Endocarpon pusillum,并首次将其细分为四个不同的垂直取样部位,分别是地上的地衣部分(L)、地下的假根部分(R)、结皮土(CS)和流沙土(S)。然后,我们从每个取样部位扩增16S rDNA的V4高变区,在Illumina NovaSeqPE250平台测序后分析。本研究提出以下科学假设:地衣可以从环境中有针对性地选择与其共存的微生物,以维持其自身的稳态,而这种稳态很少受到地衣物种的影响。为此相应地设计了以下研究目标:1)解析石果衣结皮不同部位的细菌群落组成,2)阐明影响结皮细菌群落构建的因素,3)探索其可能的来源和潜在功能;4)揭示结皮中细菌组的分子共存网络结构差异。
结 果
Illumina测序数据整理的类群划分
本研究一共120份样品,包括卷边石果衣(E. adsurgens)66份和石果衣(E. pusillum)54份,一共13,058,562条高质量序列。去除重复序列和读数小于 8 条的测序数据后,降噪生成了22,808个ZOTU。去除1156个不属于细菌界的ZOTU后,最终属于21,652个ZOTU的6,682,292条读数用于后续分析。由于120个样品中的读数范围从5717到309,940不等,因此按照每个样品5717条序列抽平,最终共686,040条序列。在S、CS、R和L四个取样部位特有的ZOTU数分别为376、296、681和160个,四个部位共有的ZOTU为10,247个(附图1)。
石果衣结皮中细菌群落多样性和构建过程
Kruskal-Wallis检验结果显示石果衣结皮取样部位分组对细菌群落的香农指数有显著影响(χ2 = 92.419,p < 0.001),采样点(χ2 = 3.0246,p = 0.2204)和石果衣物种(χ2 = 1.4674,p = 0.2258)的影响不显著。同样,取样部位(χ2 = 83.944,p < 0.001)、采样点(χ2 = 5.7553,p = 0.05627)和石果衣物种(χ2 = 2.4179,p = 0.12)对丰富度指数的影响结果类似。这两个α多样性在三个采集地点间都呈从S、CS和L到R逐步下降的趋势(Dunnett检验,p < 0.05)(图1A)。这些结果说明,土壤样品(S和CS)中细菌物种多样性较高,而地衣样品(L和R)中细菌物种多样性较低。
Bray–Curtis距离的CPCoA分析可知,四个取样部位具有不同的细菌类群组成,地衣样品(L和R)与土壤样品(S和CS)具明显差别(p =0.001,图1B)。不同采样点的细菌组成之间也存在显著差异(adonis,p = 0.037),但不同取样部位对群落差异的解释量更大(R2stratum = 0.081,R2site = 0.028)。PerMANOVA检验细菌群落在石果衣的两个物种间组成不存在显著差异(p = 0.076)(图1B),这支持了本研究提出的科学假设,即石果衣微生物群落组成不受地衣物种的影响,此后两种石果衣的样品合并分析。尽管三个采样点相距数千公里,但L中的细菌群落紧密聚集(图1B)。β离散度分析显示,从R、S、CS到L,群落相异性逐渐降低(附图2)。距离衰减模式表明随着环境距离的增加,土壤样品(S和CS)的细菌群落周转率高于地衣样品(R和L),群落相似性的下降速率更快,环境距离对群落相似性的解释量从S(0.068)、CS(0.043)和R(0.011)到L(0.0005)降低(附图3)。
土壤S和CS中的细菌组成在门水平上略有差异(图1C),放线菌门Actinobacteriota类群占优势,平均相对丰度分别为38.5%和31.2%,其次是变形菌门Proteobacteria和酸杆菌门Acidobacteriota,分别占14.3%和11.9%、19.0%和9.9%。相比之下,R和L中高丰度门组成存在明显差异,放线菌门(32.1%)、酸杆菌门(9.78%)和疣微菌门Verrucomicrobiota(2.81%)在R中更丰富,而变形菌门(68.9%)、蓝细菌门(2.57%)和拟杆菌门Bacteroidota(4.95%)在L中相对丰度更高(图1C和1D)。
根据微生物r/K策略在门水平上划分的细菌类群相对丰度在不同取样部位间有所不同(Dunn’s Kruskal–Wallis多重比较,p < 0.05)(图1D)。寡营养类群,如酸杆菌门、放线菌门、浮霉菌门Planctomycetes、绿弯菌门Chloroflexi和疣微菌门在土壤样品(S和CS)中相对丰度更高;相反地衣样品(L和R)中,富营养类群相对丰度较高,如拟杆菌门和变形杆菌门(图1D)。相对丰度最高的35个属根据系统发育关系可分为两类,S和CS中的组成更相似,并且可以与L中的高丰度属组成明显区分。L中蓝细菌组成主要以微鞘藻属Microcleus、拟色球藻属Chroococcidiopsis和Craurococcus为主。R具有相对含量较高的植物放线菌属Actinophytocola、德沃斯氏菌属Devosia、芽孢杆菌属Bacillus和克雷伯杆菌属Kribbella。
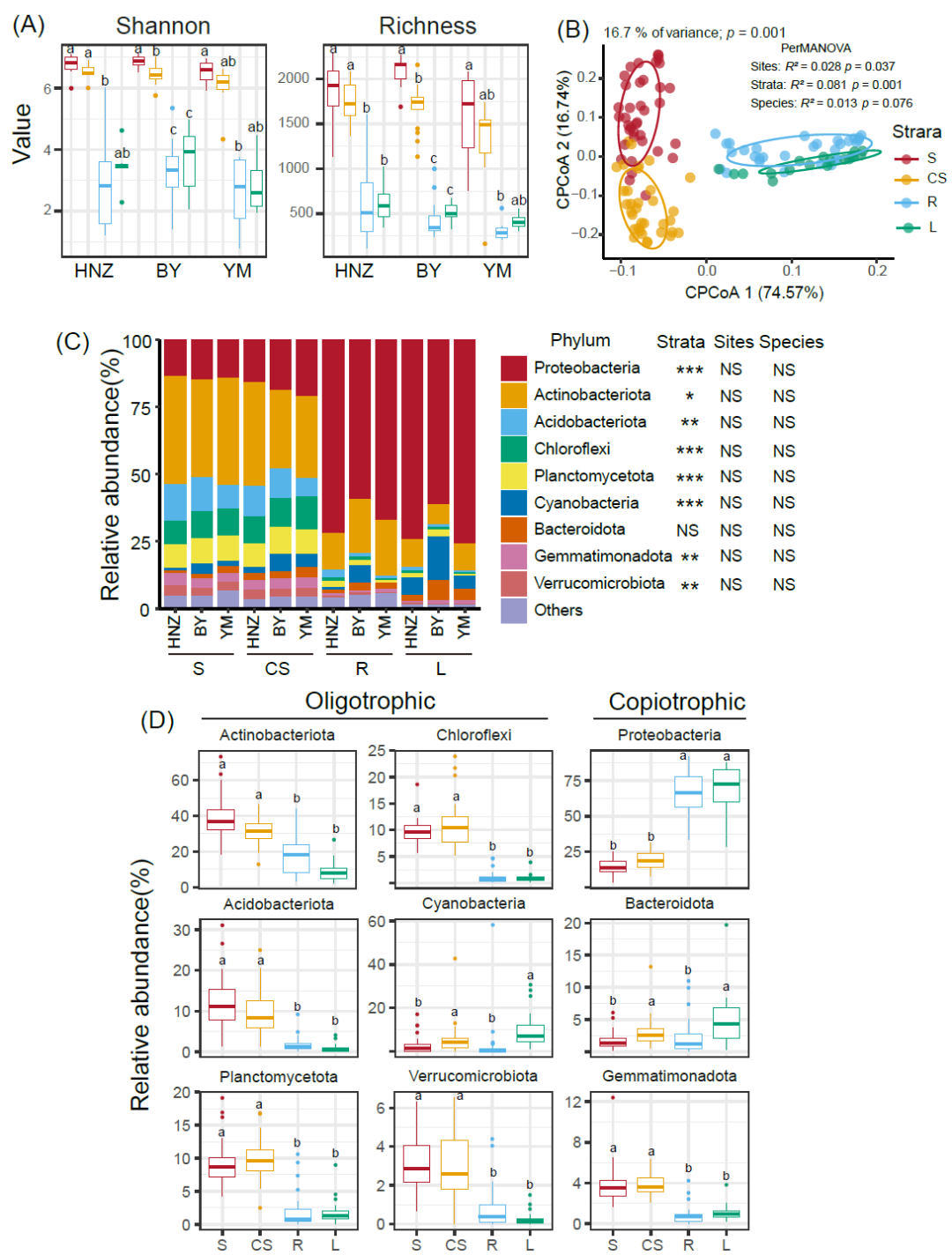
图1. 石果衣结皮细菌物种多样性和组成
(A)以丰富度指数和Shannon指数显示不同取样部位的细菌α多样性;(B)基于Bray-Curtis距离的限制性主坐标分析(CPCoA);(C)不同样品中相对丰度排名前九的细菌门组成图;(D)四个取样部位的细菌在门水平按照寡营养与富营养类群划分的组成。
典型相关分析结果,采样点年平均气温(MAT)、年平均降水量(MAP)和海拔高度可以解释4.03%的细菌群落差异,Monte Carlo置换检验表明MAP和MAT显著影响细菌群落组成(p < 0.001)。CCA分析第一维(CCA 1)能明显区分HNZ和其他地点的样品,第二维(CCA 2)区分开BY和YM样品(图2A)。方差分解分析结果显示取样部位解释了石果衣细菌群落20.6%的差异,大于环境因素(MAP和MAT,总解释量0.83%)和空间距离(PCNM1和PCNM2,总解释量0.52%)(图2B)。
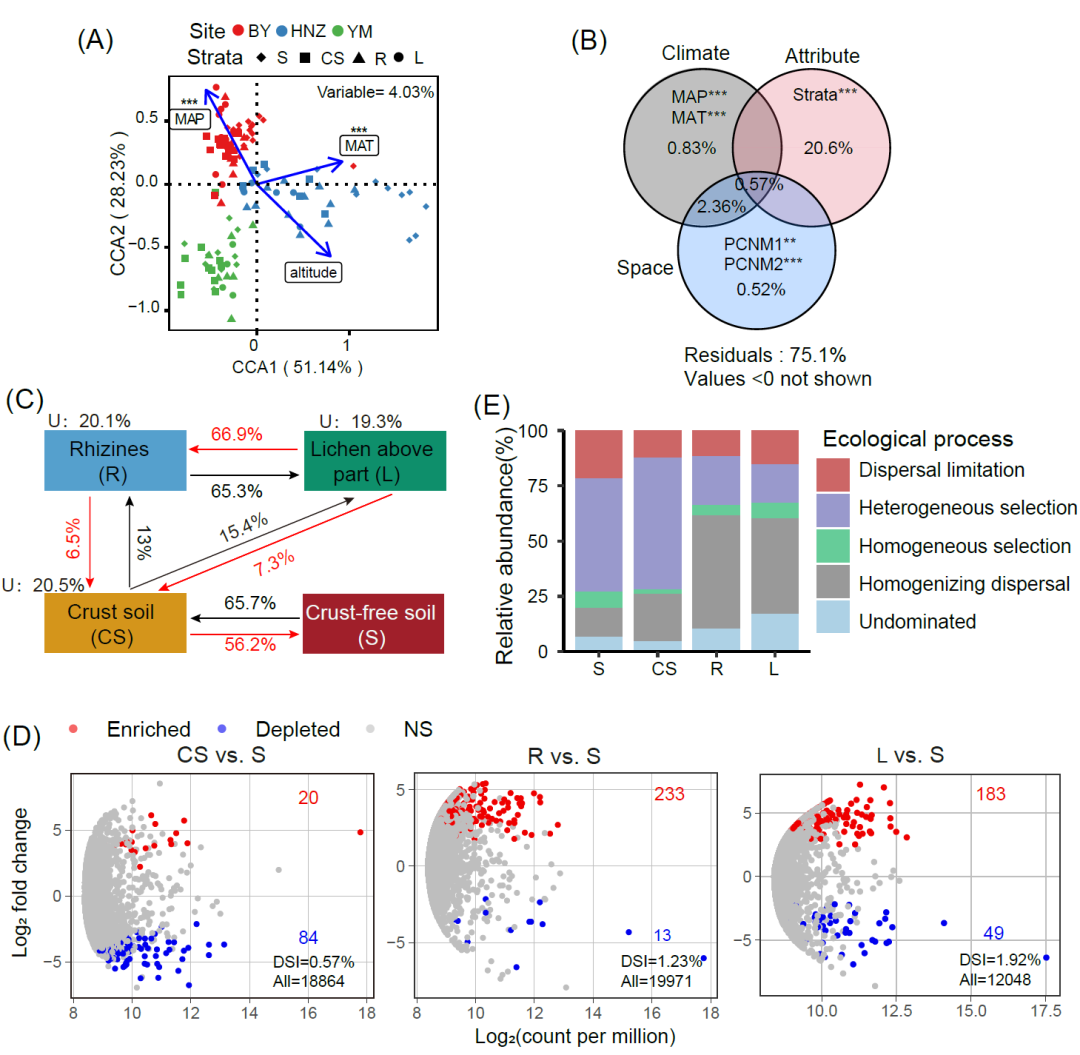
图2. 石果衣中细菌群落受环境因素、来源和组装过程的影响
A)细菌群落组成与已知环境因素的典型相关分析;(B)气候因子、取样部位和空间距离对细菌群落差异解释量的Venn图;(C)溯源分析显示石果衣细菌群落的潜在来源;(D)与S相比各取样部位细菌群落富集和衰减模式的火山图;(E)四个取样部位的划分的群落构建五个过程的相对重要性。
具宿主选择倾向的微生物群逐级选择
由溯源分析获知与地衣(L和R)相关的细菌群落主要来源于结皮土,并在垂直取样部位中逐级过滤。L直接从R中获得的主体类群主要是从CS富集而来,S是CS的物种库(图2C),说明地衣可以从环境中有选择性地选择与其共存的微生物群。EdgeR分析结果显示L富集了比例最高(183/12,048)的ZOTU,CS筛减了比例最高(84/18864)的ZOTU(图2D)。属于嗜热菌纲Thermoleophilia的ZOTU在R(48/233)和L(41/183)样品中显著富集,但在CS(16/84)中显著筛减;而α变形菌纲Alphaproteobacteria在CS(16/20)样品中明显富集,但R(6/13)和L样品中显著筛减(详见附表1)。差异指数(DSI)从CS(0.57%)到R(1.23%)和L(1.92%)逐渐增加,表明地衣宿主对细菌群落的选择作用在垂直取样部位中自下而上逐渐增加(图2D)。
石果衣结皮的群落构建过程分析在四个取样部位中结果不同。地衣样品中的随机过程(扩散限制、同质扩散和非主导作用)重要性增加,在R(73%)和L(75%)中的随机过程主要为同质扩散(51%和43%)。相反,确定性过程(异质选择和同质选择)对土壤样品中细菌群落构建的影响更大,S(59%)和CS(62%)中的确定过程主要为异质选择(均超过50%)(图2E)。
取样部位影响细菌共存网络拓扑结构和关键物种
共存网络的平均路径长度、平均度和平均聚类系数都高于Erdös–Réyni随机网络(附表2),表明细菌组的共存关系是非随机的。网络的拓扑结构在四个取样部位间有明显的变化(图3A,附表2,Kruskal–Wallis检验)。具体而言,S具有最高的网络连通性(即网络度、紧密中心性、中介中心性、特征向量中心性和连通性)(p > 0.05)。细菌网络对干扰的抵抗力通过去除随机节点进行评估,当去除50%的随机节点时,剩余节点数目表示网络鲁棒性。S网络表现出显著高于其余部位的鲁棒性(p > 0.05)。相比之下,L网络具有仅次于S中网络的连通性(节点的度)、中心性和复杂性(连通性)(p > 0.05)。此外,L网络的模块性最低,其中的细菌群落更为复杂和高度连接,有可能与地衣共生体稳态维持有关。R网络最简单,并且脆弱性最高(图3B)。四个取样部位的网络都以正相关为主,从CS(10.36%)、S(5.31%)和L(0.93%)到R(0.88%),负相关比例呈逐步下降趋势(Dunnett检验,p < 0.05)(附表2)。每个网络中的大多数节点都是外围节点,关键节点主要是连接节点。统计网络这关键节点所属的门在四个取样部位中各不相同(附表3)。在S网络中,连接节点属于α变形菌纲、拟杆菌纲和放线菌纲,网络核心节点属于假诺卡菌科。L中的连接节点属于16个纲(嗜热油菌纲Thermoleophilia、红杆菌纲Rubrobacteria、蓝细菌纲Cyanobacteria等)。CS网络中的模块中心节点属于拜叶林克氏菌科Beijerinckiaceae,R网络中的模块中心节点属于纤线杆菌纲Ktedonobacteria、芽孢杆菌目Bacillales和鞘氨醇单胞菌目Sphingomodales(图3C,附表3)。
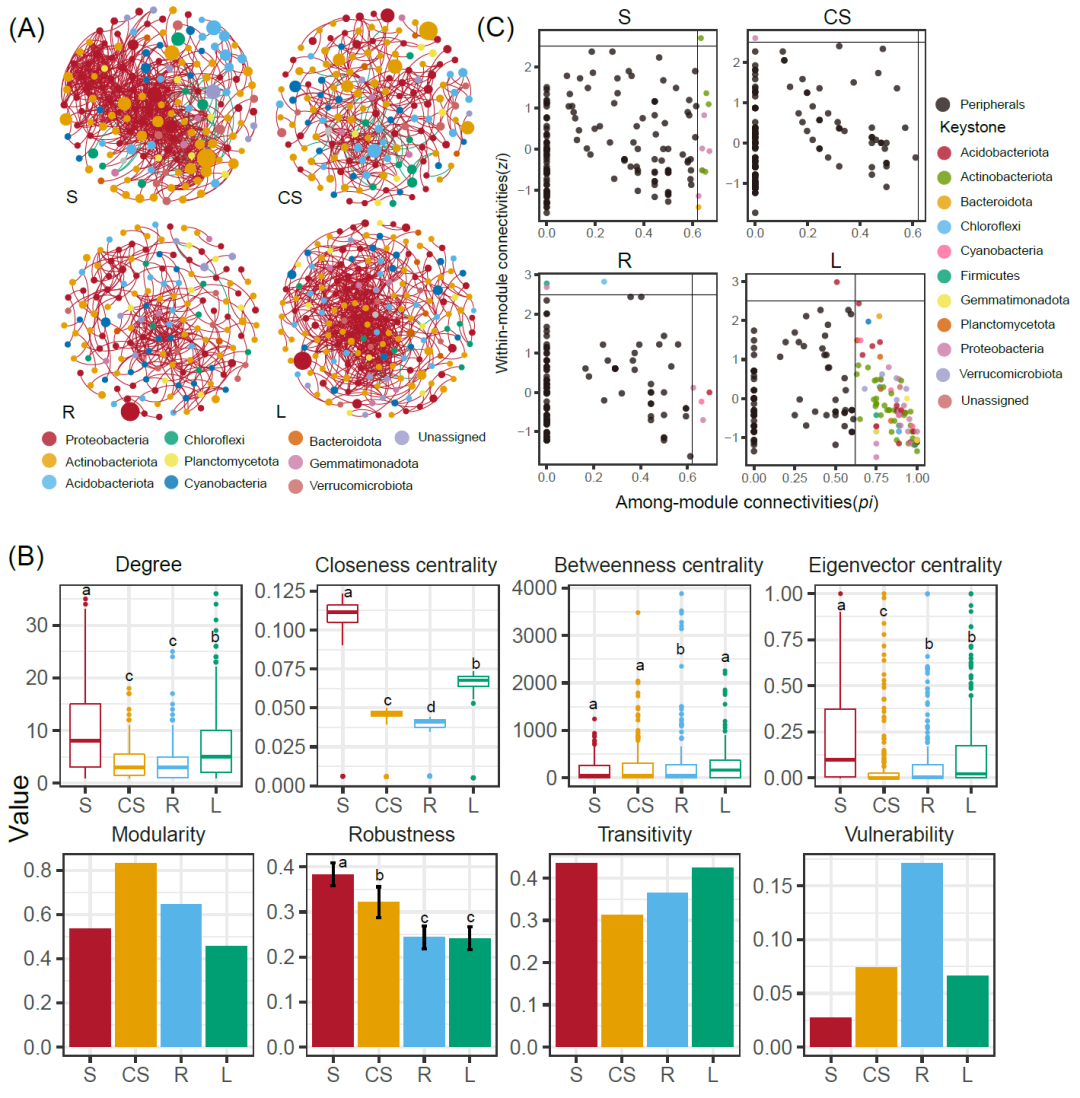
图3. 四个取样部位间细菌群落的共存网络、网络基石节点和网络拓扑结构
(A) 四个取样部位细菌共存网络概览。红色链接表示正相关,绿色链接表示负相关;(B) 网络节点和网络整体的部位拓扑结构;(C) 四个取样部位网络中的潜在关键节点。
方 法
根据年平均降水量MAP、年平均气温MAT和采样点海拔,本研究选择了差异较大的荒漠进行样品采集:海南藏族自治州HNZ、甘肃省白银市BY和玉门市YM。HNZ位于青藏高原东北部,平均海拔大于3000米。YM位于甘肃省西北部,地形为山地、平原和戈壁。BY地处黄土高原向腾格里沙漠的过渡地带。采样时使用无菌铲收集样品,分别储存在无菌聚乙烯袋中,并在干冰上冷却,低温运送到实验室进行下一步处理。所有采样点的MAP和MAT均来自WorldClim数据库(www.WorldClim.org)。将石果衣结皮细分为四个取样部位(详见附图6),包括1)地衣地上部分(L),指的是去除假根后的地衣体裂片;2)地衣地下假根部分(R),指地衣体下表面的假根;3)结皮土(CS),指L下方5 mm处假根周边附着的沙土,以及4)流沙土(S),指的是L下方5 cm处的无结皮沙土。采样点和样品的详细信息参见附件材料附图7、附图8和附表4。
土壤样品(CS和S)在研钵中研磨并通过0.2 mm筛子除去植物碎屑和砾石。将约0.5 g土壤样品直接加入PowerSoil DNA提取试剂盒(MoBio Laboratories Inc., Carlsbad, CA, USA)中,按说明书提取基因组DNA。地衣样品(L和R)通过75%乙醇漂洗1分钟,并用无菌水漂洗三次,进行表面消毒,采用改良的CTAB方法提取基因组DNA。
在25 µl反应体系中扩增16S rDNA的V4高变区,反应体系包括2.5 µl 10× Ex Taq缓冲液(20mM Mg2+),上下游引物各0.5 µM,2µl dNTP(每种2.5 mM),0.5 U Ex-Taq(TaKaRa),以及10 ng模板DNA。使用的引物是515F和806R,引物包含12碱基的条形码序列。PCR扩增包括95°C预变性5分钟,接着30个循环:95°C变性30秒,56°C退火40秒,72°C延伸40秒,最后72°C延伸5分钟。每个PCR产物三个重复,在1%琼脂糖凝胶上电泳检查无误后混合并纯化。使用NanoDrop One分光光度计(Thermo Scientific, Wilmington)检测纯化产物的质量,并使用Qubit2.0(Thermo Scientific, Wilmington)精确测量核酸浓度。最终将相同摩尔量(100 ng)的纯化PCR产物混合。构建测序文库,并在Illumina NovaSeq PE250平台上进行测序。本文中的原始序列数据存储在NCBI数据库PRJNA873937项目中。
所有样品的双向序列通过USEARCH 10中的fastq_mergepairs合并,fastq_filter命令进行质量过滤,然后对样品进行重命名并去除引物。本研究使用USEARCH中的uchime命令根据SILVA数据库(版本138)检查和删除潜在的嵌合体序列。剩余的序列中去除冗余序列,并且丢弃低丰度(总计数<8)序列。使用默认参数对这些高质量的非嵌合体序列进行unoise2降噪,得到的ZOTUs使用SILVA数据库(版本138),根据置信度阈值0.8进行分类鉴定。去除注释到叶绿体、线粒体或古菌的ZOTUs。
所有下游的统计分析均在R(版本4.0.5)中进行(HTTP://www.r-project.org)。使用USEARCH计算α多样性指数(Shannon指数和丰富度指数),并用非参数统计检验(Kruskal-Wallis检验,Dunnett's检验)不同采样点和取样部位的样品之间的差异。限制性主坐标分析(CPCoA)检验Bray–Curtis距离矩阵的群落间相似性,使用vegan包中的adonis函数进行PerMANOVA检验999次测试确定分组方法对群落差异的影响程度。距离衰减关系计算细菌群落使用Bray–Curtis指数,环境距离转换为欧氏距离。本研究选择了相对丰度最高的九个门,其余的低丰度门类划分到为“其它”,通过堆积柱状图展示物种组成,使用Kruskal-Wallis检验采集地点、取样部位和物种之间对高丰度门组成差异影响。同时,根据细菌利用资源的方式划分为寡营养型和富营养型,比较在四个取样部位间的组成差异。典型相关分析(CCA)使用Monte Carlo置换(999次重复),检验环境因子(MAP、MAT和海拔)对细菌群落结构的影响。使用vegan包中的varpart函数进行方差分解分析(VPA),确定已知变量对细菌群落差异的相对贡献,采集点之间的空间距离通过使用pcnm函数转换为地理坐标的欧氏距离。采用PerMANOVA检验气候因子(MAP和MAT)、样本属性(取样部位)和空间距离对细菌群落组成的影响。
使用SourceTracker包(1.0)来确定每个取样部位细菌群落的潜在来源。使用EdgeR广义线性模型方法对ZOTU差异分析(富集:FDR < 0.05,log2FC >= 1,筛减:FDR < 0.05,log2FC <= 1,无差异:FDR >= 0.05或|log2FC| > 1),并定义差异指数(DSI)来评估从潜在物种库到其他取样部位的细菌选择过程。用Raup-Crick指数(RCI)和βNTI(beta最近分类单元指数)计算每个取样部位中细菌群落构建过程。
在不同的取样部位间分别构建了微生物共存网络比较群落互作模式的差异,数据集选择相对丰度 > 0.05%的ZOTU,选择阈值是Spearman相关系数(r值)> 0.6和FDR校正p值 < 0.05,使用igraph包构建网络。计算以下网络拓扑结构:平均度、平均路径长度、直径、紧密中心性、介数中心性、特征向量中心性、模块性、连通性、鲁棒性和脆弱性。根据节点在模块内连接性(Zi)和模块间连接性(Pi)划分网络的关键节点。网络使用交互式平台Gephi可视化。
讨 论
本研究中从土壤到地衣样品,细菌群落的α多样性逐渐降低(图1A),由于目前缺乏关于地衣结皮不同取样部位的细菌群落α多样性的研究报道,尚无法将本研究结果与现有的地衣文献进行比较。稍微相关的研究报道了地衣细菌在不同生长阶段、地理区系和生长基质上的群落结构变化。尽管细菌组被认为对地衣共生体非常重要,但迄今为止,只对地衣的上下皮层进行过显微观察。有一项早期的研究显示地衣体影响了其生长基质岩石中的细菌群落。通过细分石果衣结皮垂直取样部位,本研究填补了这些研究之间的空白,并首次评估了各取样部位间细菌群落组成的差异。
细菌组在不同环境距离之间的群落相似性降低,表明存在距离衰减的模式。土壤样本中,β多样性的距离衰减曲线的斜率更高(附图3),表明土壤样本具有更高的分类和系统关系周转率,这可能是由于地衣共生体对细菌群落产生了强烈的宿主选择效应,以维持地衣共生体中相对稳定的微生物群。本研究结果很好地支持了先前提出的科学假设,即地衣可以从环境中有针对性地选择与其共存的微生物群,以维持其自身的稳态,而且这种稳态关系很少受到地衣物种的影响。
地衣可以代表具有不同功能的细菌多样化形成的生态位,物种之间的共生关系可以充分利用有限的资源进行代谢活动。富营养型物种通常具有较高的rRNA操纵子拷贝数和较高生长率,寡营养类群则具有较慢的生长速度和更稳定的种群结构。本研究结果表明,土壤样品中富集了寡营养型细菌,而地衣样品富集了富营养型细菌。在R部位富集的某些细菌类群,如芽孢杆菌(厚壁菌门),可能参与土壤中有机物质的降解。L部位的细菌种类组成与先前报道的肺衣Lobaria pulmonaria等树生地衣中的细菌种类组成相似,均是以变形菌门为主(图1C)。此外,还富集了拟杆菌门类群,这些细菌一般生长在碳源丰富的环境中。因此地衣中富营养细菌的相对丰度高可能与地衣体中容易利用的资源丰富有关(图1D)。
本研究创新性地发现,石果衣结皮四个取样部位的细菌群落构建过程不同。异质选择(确定性过程)在细菌群落构建中发挥着主导作用,有可能导致群落之间的结构差异更大。地衣样品具有空间异质性,相关微生物群会受到宿主(共生菌和光合共生物)和环境的影响,塑造细菌群落结构的高度变化。地衣共生菌和光合共生物形成的二元系统可能解释了一定比例的扩散过程,同质扩散(随机过程)会让群落之间差异更大。
L共存网络中的连接节点属于放线菌门Actinobacteriota中的土壤红杆菌目Solirubacterales、假诺卡氏菌目Pseudonocardiales和红色杆菌目Rubrobacterales,酸杆菌门Acidobacteriota中的Pyrinomonadales,以及变形菌门Proteobacteria中的根瘤菌目Rhizobiales(图3B)。R与L紧密相连,相对优势的连接节点是变形菌门中的鞘氨醇单胞菌目和根瘤菌目,它们也是在组成上丰度最高的类群。根据先前对可培养微生物的研究,大多数放线菌适应干旱条件,对干燥和寡营养条件具有高度抵抗力。最近的研究表明,放线菌在干旱胁迫期间富集,改善了植物的耐旱性并促进生长。石果衣通常生长在干旱、半干旱的荒漠中。因此,我们推测R部位富集的放线菌门可能具有类似的功能,例如通过帮助地衣利用基质中的营养物质来促进地衣的生长。其他一些与地衣相关的细菌(详见附表3)可能有助于地衣抵御环境胁迫,例如鞘氨醇单胞菌目内有可光合作用的类群,根瘤菌目已知可与植物形成固氮共生关系和参与地衣中特定次生代谢产物和营养循环。根瘤菌目中的拜叶林克氏菌科Beijerinckiaceae和黄杆菌科Xanthobacteriaceae在地衣的R和L部位均富集。黄杆菌科是好氧化能异养型,科内类群普遍具有固氮功能,而拜叶林克氏菌科内的大多数类群会产生荚膜多糖,能够固定二氮化合物。因此,我们推测这些存在于地衣中的细菌可能并非随机偶然现象。此外,由于地表下取样部位获取的光照有限,R中丰富的鞘氨醇单胞菌目暗示这里可能存在另一种营养获取途径,鞘氨醇单胞菌内包含很多好氧不产氧型光能异氧细菌,具有多样的类胡萝卜素和光合作用基因簇(PGCs)。包括石果衣在内的绿藻地衣获取氮的来源尚不完全清楚,因此,L中高丰度的根瘤菌引起了我们的注意,它们在其它绿藻地衣中也普遍存在,然而,没有证据支持根瘤菌目细菌像在植物中一样有助于绿藻地衣的固氮。本研究还发现,L中的蓝细菌数量相对较高,束鞘藻科Coleofasciculaceae、色球藻科Chrooccciopsaceae、席藻科Phormidiaceae和念珠藻科Nostoccaceae被鉴定为网络连接节点(详见附表3),它们在绿藻地衣共生中是否发挥固氮作用值得进一步探究。
代码和数据可用性
文中使用的原始序列数据已存放于GenBank下的BioProject中,登录代码为PRJNA873937(https://www.ncbi.nlm.nih.gov/sra/?term=PRJNA873937)。附件材料(方法、图、表、脚本、图形摘要、幻灯片、视频、中文翻译版本和更新材料)可在线获取。
引文格式:
Zhang, Tingting, Martin Grube, Xinli Wei. 2023. “Host selection tendency of key microbiota in arid desert lichen crusts”. iMeta e138. https://doi.org/10.1002/imt2.138
作者简介

张婷婷(第一作者)
● 张婷婷,中国科学院微生物研究所硕士研究生,现中国科学院动物研究所博士生在读。
● 研究方向为地衣结皮微生物组和地衣型真菌分类。

魏鑫丽(通讯作者)
● 中国科学院微生物研究所研究员,中国科学院青年创新促进会会员。
● 研究方向为陆地先锋生物地衣共生体系的认知、构建及资源应用,已在Nature Communications、PNAS、eLife等期刊录用和发表学术论文70余篇,主编出版专著1部,主持国家自然科学基金项目、科技部基础资源调查专项课题、北京市自然科学基金等17项,获西藏自治区科技进步一等奖和北京市自然科学奖二等奖。
更多推荐
(▼ 点击跳转)
iMeta | 引用7000+,海普洛斯陈实富发布新版fastp,更快更好地处理FASTQ数据
iMeta | 德国国家肿瘤中心顾祖光发表复杂热图(ComplexHeatmap)可视化方法


1卷1期

1卷2期

1卷3期

1卷4期

2卷1期

2卷2期

2卷3期

2卷4期
期刊简介
“iMeta” 是由威立、肠菌分会和本领域数百位华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表原创研究、方法和综述以促进宏基因组学、微生物组和生物信息学发展。目标是发表前10%(IF > 15)的高影响力论文。期刊特色包括视频投稿、可重复分析、图片打磨、青年编委、前3年免出版费、50万用户的社交媒体宣传等。2022年2月正式创刊发行!
联系我们
iMeta主页:http://www.imeta.science
出版社:https://onlinelibrary.wiley.com/journal/2770596x
投稿:https://mc.manuscriptcentral.com/imeta
邮箱:office@imeta.science

















 360
360

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









