






微生物富集方法使得从富含宿主的样本中进行高通量宏基因组表征成为可能
Microbial-enrichment method enables high-throughput metagenomic characterization from host-rich samples

Article,2023-10-12,Nature Methods, [IF 48]
DOI:https://doi.org/10.1038/s41592-023-02025-4
原文链接:https://www.nature.com/articles/s41592-023-02025-4
第一作者:Natalie J. Wu-Woods, Jacob T. Barlow
通讯作者:Rustem F. Ismagilov
合作作者:Florian Trigodet, Dustin G. Shaw, Anna E. Romano, Bana Jabri, A. Murat Eren
主要单位:
加州工学院(Biology and Bioengineering, California Institute of Technology, Pasadena, CA, USA. Department of Medicine)
芝加哥大学(The University of Chicago, Chicago, IL, USA)
Committee on Immunology, The University of Chicago, Chicago, IL, USA.
Department of Pathology, The University of Chicago, Chicago, IL, USA. Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, USA.
Bay Paul Center, Marine Biological Laboratory, Woods Hole, MA, USA.
德国奥尔登堡大学(Institute for Chemistry and Biology of the Marine Environment, University of Oldenburg, Oldenburg, Germany)
Alfred-Wegener-Institute for Marine and Polar Research, Bremerhaven, Germany. Helmholtz Institute for Functional Marine Biodiversity, Oldenburg, Germany.
- 摘要 -
目前,主要通过基于16S核糖体RNA基因测序的微生物分类鉴定,已经确定了宿主与微生物间的相互作用与宿主健康和疾病有紧密关联,但许多关键机制仍待揭示。其中一个难点是,在宿主DNA与微生物DNA比例极高的样本中,研究与哺乳动物组织关联的微生物基因组异常困难。在本研究中,我们提出了一种名为MEM(microbial-enrichment method;MEM)的微生物富集方法,并对其在多种类型的样本上进行了验证,如唾液、粪便、肠道刮片和肠黏膜活检。MEM通过在微生物群落变化最小的情况下(大约90%的微生物种类在处理与未处理样本间无显著差异)将宿主DNA减少1000倍以上,实现了对人类肠道活检微生物宏基因组的高通量表征。随机(Shotgun)宏基因组测序经过MEM处理的人类肠道活检样品能够表征沿胃肠道纵向的高丰度和低丰度微生物分类群、代谢通路和功能基因。此外,我们还从人类肠道活检中成功构建了相对丰度低至1%的细菌和古细菌的宏基因组组装基因组(metagenome-assembled genomes;MAG)。进一步对宏基因组组装基因组的分析揭示了某些微生物在小肠与大肠间具有不同的亚群结构。MEM技术为微生物组学领域开辟了新的途径,使我们能够更深入地理解与宿主组织相关的微生物群落,从而更深刻地揭示宿主和微生物之间的相互作用。
- 引言 -
肠道黏膜上的微生物群落与包括癌症、炎症性肠病(IBD)以及乳糜泻在内的众多健康问题息息相关。尽管大便和肠道活检中的微生物有着不同的生态位,但由于采样方便,大多数微生物组学研究多使用粪便样本来推测胃肠道(GI)中的微生物。而且大部分都采用16S核糖体RNA (rRNA)基因扩增子的测序来描述微生物群落分类。近年来,由于宏基因组测序能够提供更为深入的微生物基因组特征而被广泛应用于人类微生物生态学研究。此外,宏基因组测序还可以解析单一分类群内的亚种结构,例如生理上的宿主梯度如何在微生物组上施加进化压力,并对研究哪些微生物基因在不同宿主环境下受到选择压力均具有重要作用。
人们对组织相关微生物如何与宿主环境相互作用的分子机制仍缺乏足够的了解,因为该领域缺乏相称的工具来超越分类解析并直接从肠道活检样本中研究微生物代谢通路和功能基因。全基因组表征的两种常见方法包括培养微生物分离物或直接从混合的微生物样本中重构宏基因组组装的基因组(MAG)。依赖培养的方法有一定作用;然而,独立于培养的方法对于从其原始环境中描述微生物,以及那些难以分离的微生物而言具有重要价值。MAG是通过测序及先进的算法创建的,其中测序读序被组装成连续的序列,然后被分组到单独的分箱(bins)中,以在不培养的情况下重构完整的基因组。
宏基因组测序对复杂的宿主相关微生物组的分析受到了样品中存在的宿主与微生物核酸高比率的挑战。在人类样品中,唾液样本中的85-95%读序是宿主的,而肠道活检中超过99.99%是宿主的。这些宿主与微生物DNA的巨大比率(在人肠道活检中为1:10,000)对于宏基因组测序研究提出了巨大挑战,因为大多数测序序列与宿主基因组一致。直接使用当前的分析方法和测序深度对这样的组织样本无法产生足够的微生物测序序列来构建MAG。
为了防止大多数的宏基因组测序序列分配给宿主,已经开发了各种各样的宿主去除(host-depletion)方法。已发表的和商业的方案已经使得从哺乳动物源液体样本中进行长读序测序和细菌MAG构建成为可能。尽管一些方案已经使用固态组织类型的样本进行了验证,而其他方案可能在这些样本中有成功的案例,但没有任何一个方案被证明能足够有效的从固态哺乳动物组织中构建细菌MAG。此外,许多宿主去除的方法由于处理时间过长和方案复杂而不适用于临床操作。
该研究开发和优化了一个微生物富集方法(MEM),以从复杂样本中去除宿主核酸,而不大幅度扰乱微生物群落的组成。研究人员在实验室和临床环境中展示了MEM的性能,包括唾液、粪便、肠道刮片和肠道活检在内的各种样本类型。还展示了MEM后续的宏基因组测序在沿着胃肠道的人类肠活检中检测高丰度和低丰度微生物分类群、代谢通路和基因的能力。此外,研究人员还对使用MEM直接从人类肠活检中构建MAG,以识别和区分亚种群和亚种群变种优异性能进行了展示。
- 结果 -
MEM能最大限度地减少样品处理过程中的细菌损耗
MEM minimizes loss of bacteria during sample processing
MEM通过利用宿主细胞和细菌细胞之间的大小差异,结合了使用机械压力(bead beating)的选择性裂解方案从而选择性的裂解宿主细胞和细菌细胞(Fig.1a,b)。通常用于微生物裂解的珠子大小为0.1-0.5mm,我们选用更大的珠子(1.4mm)裂解更大的宿主细胞同时保持小的细菌细胞完整性。随后添加核酸酶降解胞外核酸和裂解死亡的微生物核酸。蛋白酶K进一步裂解宿主细胞并降解宿主组蛋白以释放DNA。研究人员还测试和优化了影响MEM性能的其他因素,包括酶解法去除核酸,珠磨法和不同孵育时间,使整个方案保持在20 min以内,并在温和的处理条件下防止微生物裂解。为了比较MEM与现有的去宿主方法,我们选择了三种已发表的不同细胞裂解方法:MolYsis, QIAamp和lyPMA。所有的去宿主方法包括两个主要步骤:选择性裂解,然后是核酸去除。QIAamp通过一种弱清洁剂(皂苷)来裂解缺乏细胞壁的细胞。MolYsis通过暴露于低浓度的胍盐来选择性裂解更脆弱的哺乳动物细胞。lyPMA通过渗透裂解哺乳动物细胞,并利用光化学使DNA结合单叠氮丙啶(PMA)使其不能被扩增。
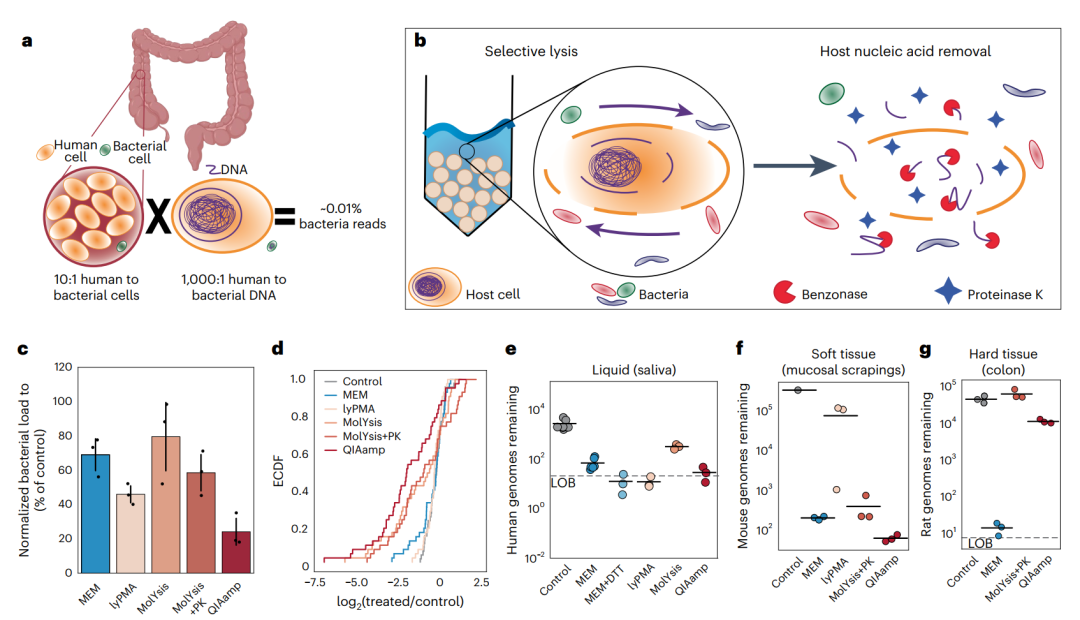
图1 MEM与已发表方法的比较。
a. 当人类肠道活检未经处理测序时获得的细菌读序的百分比。
b. MEM中使用的两步选择性裂解和核酸去除技术示意图。
c. 用五种不同的去宿主方法处理的小鼠粪便样本的细菌载量。载量根据对照样品(未经过宿主去除的粪便样本)进行了标准化(n = 3;误差为95%置信区间)。
d. 将小鼠粪便样品标准化为对照粪便样品的16S rRNA基因扩增子测序结果的经验累积分布函数(ECDF)。向左移动的曲线表明去宿主后低丰度类群比例高于对照样本。
e-g. 剩余的宿主DNA通过单拷贝宿主特异性引物ddPCR进行定量。报告的基因组剩余是指该单拷贝基因在1µl 洗脱液中存在的丰度。
e.在用每种去宿主方法处理后和在未处理的对照中对新鲜人类唾液中的剩余人类基因组进行量化。
f. 宿主去除方法在作为软组织代表的小鼠肠黏膜刮片上进行了测试,并对剩余的小鼠基因组进行了量化。
g. 宿主去除方法在作为代表性硬组织(包括结缔组织、肌肉和黏膜)的大鼠结肠切片上进行了测试,并对剩余的大鼠基因组进行了定量。
为了量化MEM如何影响微生物群落的组成和单个分类群的相对丰度,我们首先使用冷冻小鼠粪便样本。我们选择粪便样本而不是人工合成菌群来表征微生物对一系列独特分类群和连续丰度的影响。这是因为小鼠粪便样本通常不需要去除宿主,因为它们的宿主污染较低(超过90%的DNA来自非宿主细胞)。微生物细胞的高含量使粪便成为表征不同去宿主方法对微生物群落组成影响的理想材料。此外,由于提取试剂盒效率的变化,仍需人工合成菌群作为DNA提取的对照样品。
虽然无法提取样品中的所有DNA,但所有样品去宿主后都使用相同的提取试剂盒处理以标准化试剂盒和/或裂解效率。在均质粪便样本,我们观察到五种去宿主方法与对照组未经处理的样品相比均有类似的微生物损耗(图1c)。MEM产生了平均31%(+11%; standard deviation)的细菌损耗,而其在粪便中10%至50%的死亡微生物细胞的预期比例范围内。为比较MEM和其他去宿主方法对微生物组分类水平上的影响,研究人员对小鼠粪便样本进行了16S rRNA基因的定量分析。通过比较,作者发现lyPMA和QIAamp方法对细菌总量损耗最大,而MolYsis和QIAamp的细菌损耗最不均匀,一些分类群下降超过100倍(图1c,d)。先前的研究建议QIAamp可以降低皂苷的浓度来减少这些细菌的损耗。作者证实,MEM微生物群落损耗最小;超过90%的属的相对丰度差异在MEM与对照样品中无显著性。此外,所有检测到的分类群与对照样品中一致而MolYsis和QIAamp则有缺失(附表1)。因为MEM选择性裂解是根据宿主细胞的细胞大小,而化学裂解的裂解程度可能根据细菌细胞壁和/或膜结构的不同而不同,因此,MEM与化学裂解(MolYsis和QIAamp)相比能引起更低的细菌偏好。
为确定MEM和其他方法的去宿主效率,作者对三种类型的样品(液体、软组织、实体组织)定量了宿主含量 (Fig.1e-g),其中实体组织宿主DNA占总量的99.9%。在唾液样本中都能一定程度上的减少宿主。经MEM处理后,宿主减少超过40倍(图1e)。由于唾液黏蛋白含量高而添加的二硫索糖醇(DTT)预处理略微增加了MEM对宿主的去除(图1e和附图3)。lyPMA在去除宿主方面表现得非常有效,但难以高效地使用,因为该方法的化学计量学性质在宿主水平低于预期时可能导致大量微生物损失(附图3)。此外,MolYsis显示出细菌回收率增加。可能是由于额外的突变裂解步骤造成的。
之后,作者检查了整个组织样本的去宿主,从上皮层的小鼠肠道刮样开始分离黏膜相关细菌(附图4)。MEM和一些已发表的方法均有效地去除了刮削样品中的宿主污染 (图1f)。MEM, MolYsis和QIAamp都显示出了约1000倍的宿主去除率(MEM平均消耗1600倍±170),其中,QIAamp表现出来了略大的宿主去除效果。lyPMA在软组织样品上表现不佳,因为该方法依赖于紫外线激活的交联,使其与不透明类型样品不兼容。
接下来,作者利用在解剖学上和人体肠道活检较为相似的硬组织样本大鼠结肠切片上测试了去宿主方法。由于lyPMA在软组织上的表现不佳,因此本实验中作者将其在硬组织实验中被去除(图1f)。研究发现,MEM是唯一对固体组织样本有效的方法(图1 g)。MEM的处理几乎能够完全去除宿主DNA(去除3600倍,s.d 1500),而MolYsis和QIAamp处理后的宿主DNA水平与对照组相似。
这些实验证明MEM适用于固体组织,可以将宿主DNA去除1000倍以上同时使微生物相对丰度的损失降到最低。在实验中,MEM处理和对照样品的16s相对丰度在超过90%的属中无显著差异。
MEM处理的唾液和粪便的宏基因组测序
Shotgun sequencing of MEM-treated saliva and stool
随后,作者研究了MEM在宏基因组学中的应用。由于生物量的限制,通过对对照(非宿主去除)样本进行随机法测序来准确表征微生物群落在肠道活检中是不可实现的。因此,首先使用了唾液和粪便样本来调查微生物组中与MEM处理相关的潜在偏差。作者首先证实了MEM处理能够可靠地减少小鼠粪便和人类唾液样本中的宿主序列(图2a,c)。DTT在唾液中的预处理使宿主去除效果提高了大约10倍(图2c)。
接下来,研究人员比较了对照组和MEM处理的样品测序结果。粪便和唾液中细菌类群的相对丰度与对照组的相对丰度呈高度相关(Pearson测定系数,粪便中的R2 = 0.93,唾液中MEM和MEM + DTT的R2 = 0.90;相对丰度在0.1%以上的分类群R2 = 0.93)。对于粪便和唾液样品,对照与MEM处理样品中的物种相对丰度之间的高度相关性表明,MEM并没有实质性地改变微生物组的组成(图2b、d和附表2、3)。对于唾液样品,低丰度类群与MEM样本中特定物种富集之间的相关性则不显著,通过比较低丰度物种的跨样本变异系数,可以更定量地进行研究(图2e)。MEM处理的唾液样本具有较低的变异系数(50%变异系数,95%置信区间),表明与未处理的对照组相比具有更好的可重复性。此外,MEM提高了低丰度物种的定量(相对丰度为0.05-0.5%),可以检测到在唾液中未检测到的另外10种物种。研究人员进一步证实,这些分类群不是在MEM处理过程中引入的(附图5)。这些用小鼠粪便和人类唾液样本进行的测序实验表明,MEM处理引入了最小的微生物偏差(超过98%的微生物物种的相对丰度损失不到四倍),同时在相同的测序深度检测到了额外的微生物分类群。
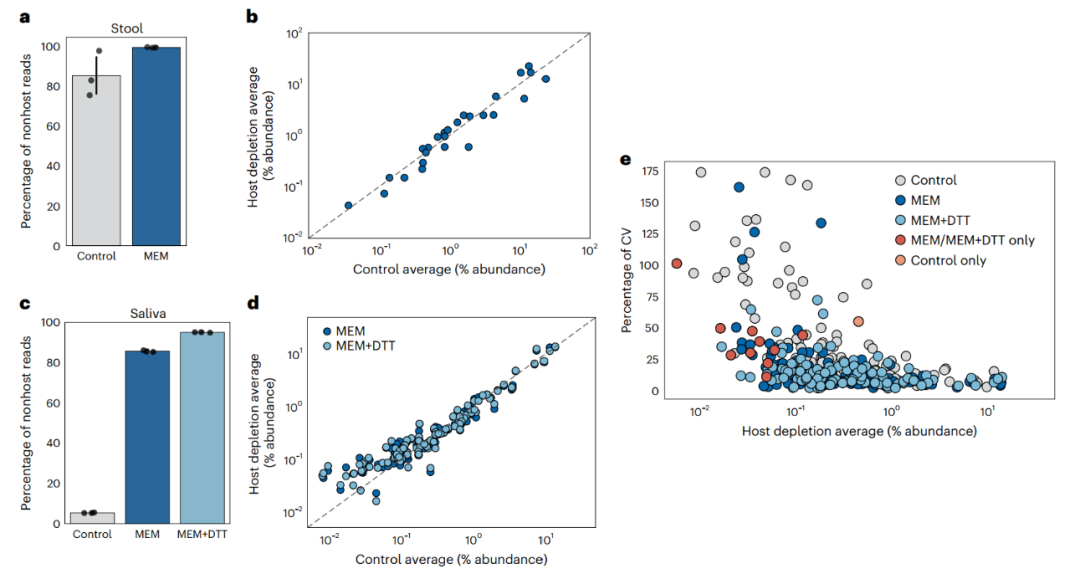
图2. 随机法测序证实MEM去除宿主后粪便和唾液的微生物富集。
a、通过与小鼠参考基因组比对,以生物信息学方法计算对照组和经MEM处理的小鼠粪便样本中非宿主读序的百分比。
b、在对照和MEM处理的小鼠粪便中绘制种相对丰度,并覆盖在显示1:1的相关性虚线上。
c、对照组和经MEM处理的新鲜人唾液进行随机测序。通过与人类参考基因组比对,以生物信息学方法计算非宿主读序的百分比。一份唾液样本被平均分成九种方式进行比较。
d、绘制了对照和MEM处理过的新鲜人唾液种水平分类群的相对丰度图,并覆盖在一个虚线上显示1:1相关性的直线。经MEM处理前进行了额外的DTT预处理的样本(MEM + DTT) 。
e、变异系数(CV)根据相对物种丰度绘制,并根据检测到的分类群的处理类型着色。每个点代表一个物种;灰色、深蓝色和浅蓝色点表示三种处理(对照、MEM和MEM + DTT)中均存在的分类群。MEM/MEM + DTT only(红点)表示仅在MEM处理样品中发现的10个分类群。仅对照(橙色点)表示仅在对照样本中发现的单一分类群,经鉴定为嗜血杆菌。
MEM在人肠黏膜活检中的可行性
MEM feasibility on human intestinal mucosal biopsies
为了确定MEM在人体肠道活检中的表现,我们招募了通过结肠镜检查进行常规结肠癌筛查的健康参与者。从4名参与者中,我们分别进行了8次黏膜活检;每位参与者的4个活检组织用于MEM的处理组和4个作为未处理的对照组(图3a)。由于担心低细菌载量样品中的污染DNA,我们还通过对MEM处理的空白进行定量16S rRNA基因测序,表征了与MEM相关的背景细菌信号和我们的处理方法(方法和附表4和5)。在所有16次活检中,MEM去除了2000多倍的宿主DNA,大多数活检的宿主水平与MEM处理后的空白相当(图3b)。
为了确定MEM如何在群落水平上影响人类肠道微生物组,我们首先进行了16S rRNA基因测序。在计算去除空白中绝对丰度较高的类群后,大约93%的属保留在了MEM处理和对照活组织检查中,这使我们相信大多数检测到的类群不是背景污染物。为了进一步证实MEM没有引入额外的污染,我们发现MEM和对照活组织检查的分类群丰度之间存在强烈的一致性(附表6)。测序结果的主成分分析表明,MEM引入的微生物相对丰度的任何差异都小于参与者之间观察到的差异(图3c)。测序结果分析显示,MEM处理后大多数类群的相对丰度变化很小,约88%的类群相对丰度与对照组没有显著差异(Mann-Whitney u检验,双侧检验P = 0.05)。在相对丰度大于1%的分类群中,95%以上的分类群在MEM和对照样品之间无显著差异。对照组和MEM处理组之间分类群的log2倍差异接近正态分布(Kolmogorov-Smirnov检验,统计量为0.074,P = 0.11)(图3d)。此外,对照和MEM处理的样品之间的相对分类群丰度呈线性相关(图3e)。当在临床人体肠道活检环境中使用时,MEM能够去除超1000倍以上的宿主污染,同时在微生物相对丰度方面引入的偏差也最小。

图3 使用和不使用MEM处理的成对人肠道活检样品中的微生物富集分析。
a、样品采集说明。
b、使用单拷贝宿主引物的ddPCR对每次活检的宿主DNA进行定量。人类基因组剩余量是指该单拷贝基因在1µl洗脱液中存在的丰度(*表示测量值低于空白,LoB的限制)。
c、活检采用16S rRNA基因测序和微生物属水平相对丰度主成分分析(PCA)来观察微生物种群变化。
d、用黑色覆盖的标准正态分布绘制了对照和MEM处理的活组织之间微生物属水平相对丰度的log2倍差异。
e、在对照和MEM处理的活组织中测量的微生物属水平的相对丰度被绘制并覆盖在虚线上,显示1:1的相关性。灰色突出显示的是低于定量限(LOQ)的分类群。橙色突出显示的分类群在对照和MEM活检之间的变化大于4倍.
MEM使研究微生物种类、代谢通路和基因成为可能
MEM enables study of microbial species, pathways and genes
为了研究MEM是否能够从人类肠道活检组织中检测和表征额外的微生物物种、功能代谢通路和基因,我们对来自CT18及其相对应的对照和MEM处理的活检组织进行了随机宏基因组测序,深度超过1亿读序(每种条件n = 2)(图3a)。研究发现与对照样品相比,MEM处理的样品中检测到的生物体数量增加了大约100倍,相关功能代谢通路数量增加了700倍,基因数量增加了400倍以上(图4a)。当只比较完整通路(定义为90%以上完成)时,在对照活检中均未检测到完整通路。对照组平均检测到1.5(±1.5)个物种和728(±107)个基因,而MEM组平均检测到137.5(±21.5)个物种和300,641(±6,922)个基因。MEM处理使宏基因测序对微生物的分类降低到相对丰度0.005%,而在对照活检中,检测类似测序深度的微生物需要最低10%的相对丰度。MEM处理的活组织可以检测到相对丰度为10−10的基因,而在对照活组织中,基因只能在最小相对丰度为10−5时被检测到。我们进一步发现,MEM处理提高了检测最丰富基因的再现性。我们比较了两个MEM处理和两个对照活检中最丰富的前5000个基因的相对丰度(图4b)。在对照活组织检查中,仅在一个样本中检测到高比例的基因(98%),而对于MEM处理的样本,仅在一个生物学重复中检测到3%的基因,而在另一个生物学重复中未检测到。
随后,我们测试了MEM是否能够在个体之间横向和纵向上跨越单个个体的胃肠道来表征微生物变异(在分类群、通路和基因水平上)。5名接受结肠镜检查的健康参与者分别在胃肠道的四个区域采样:回肠末端、升结肠、降结肠和直肠。在每个位置进行了3次活检,每位参与者总共进行了12次活检(图4c)。所有活组织切片均采用MEM处理,并通过16S rRNA基因测序和宏基因组测序对微生物分类单元进行表征,平均测序读序深度为2500万,平均产生200万非宿主读序(图4d-f和附图1和图2)。所鉴定的187种微生物中约有一半是单个个体所特有的(图4d和附表7)。这些独特物种的相对丰度范围为10%至0.01%(附图6)。正如之前观察到的,与物种分类(属和种)相比,参与者之间的通路似乎更为保守。
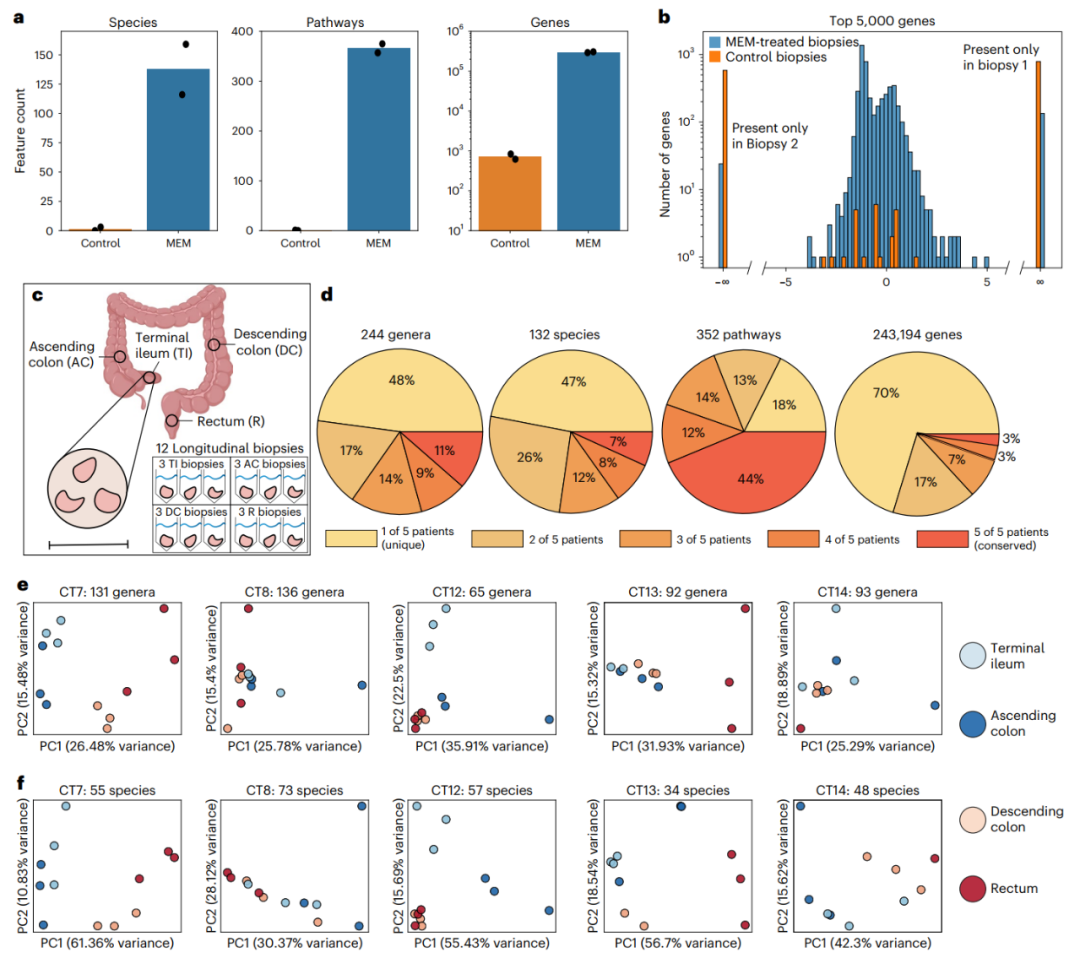
图4 经MEM处理的人类肠道活检的宏基因组测序。
a、对参与者CT18(两个MEM处理和两个对照) 的四个活检组织进行随机法测序,并绘制每个样本中鉴定的微生物种类,通路和基因的数量(n = 2)。
b,对于前5000个丰度最高的基因,绘制了两个MEM处理的活检组织与两个对照活检组织之间相对丰度的log2倍变化。
c、抽样采集示意图。比例尺(Scale bar),5厘米。
d、属、种、功能代谢通路和基因的特征数量根据它们是否存在于至少一个活检样本中,来自一个参与者,两个参与者,三个参与者,四个参与者或来自所有五个参与者。
e,f、主成分分析(PCA)对所有60个纵向样本按参与者分组。
e、16S rRNA基因测序属级别相对丰度的主成分分析。f、随机法测序物种相对丰度的主成分分析。
胃肠道黏膜微生物纵向变化
Variation in mucosal microbes longitudinally in the GI tract
由于可以从黏膜活检中获得的微生物读序较少,因此很难确定黏膜相关微生物在胃肠道中是否存在差异。本研究中,研究人员首先测试了胃肠道部位之间的微生物差异是否存在于属水平上。对于每个参与者样本,本研究使用定量16S rRNA基因测序来量化沿胃肠道纵向的属水平微生物变化。在大多数参与者中,近端结肠(末端回肠和升结肠)和远端结肠(降结肠和直肠)的微生物类群在空间位置上表现出一定的聚集性(图4e)。对每个参与者样本进行随机宏基因组测序,以测试观察到的胃肠道分类群差异是否延伸到物种、通路或基因水平。在一些跨物种的参与者中,即参与者CT7、CT12、CT13和CT14中,可以看到回肠末端和升结肠与降结肠和直肠之间的聚类(图4f)。在通路和基因水平上,回肠末端和升结肠与降结肠和直肠之间似乎存在最小的聚类(扩展数据图3)。此外,某些个体在区域内存在很大差异,这可能归因于测序深度的限制。例如,对于CT13的一个降结肠样本,由于非宿主读序的数量最少,没有鉴定出微生物标记基因(图4f)。
对经过MEM处理的人类肠道活组织进行随机测序,可以表征高丰度和低丰度的微生物物种、通路和基因。这一特征记录了沿着人类下消化道黏膜微生物组的纵向变化。为了研究单一微生物菌株是否沿着胃肠道变化,本研究也尝试了从MEM处理的肠道活检中组装微生物基因组。
MEM能够对人体活检的肠道微生物进行MAG检测
MEM enables MAG of intestinal microbes from human biopsies
为了确定MEM处理后是否可以构建MAG,本研究从参与者CT18中选择了两个具有相似细菌载量的对照和两个MEM处理的活组织切片(图3a和4a)。如前所述,对样本进行随机测序和基因组组装处理。本研究对对照和MEM处理的活组织切片进行了测序,以测量MEM处理在相同测序深度下是否能够有助于挖掘出更多信息。处理后,从生物信息学上去除宿主序列,在MEM处理的样本中,大约10%的测序读序被鉴定为非宿主,而在未经处理的对照组中,仅有大约0.01%的测序读序被鉴定为非宿主(图5a)。
本研究首先试图从对照样本中重建MAGs,然而,从非宿主去除的样本中组装短读序序列,以及我们随后试图将所得重叠群放入MAGs的尝试都没有成功,因为这些组装受到了非常短的重叠群的影响(图5b)。然后对MEM处理过的样品进行共组装,与对照样品相比,获得了更多更长的重叠群, 重叠群长度可达833 kbp(图5b和附表8)。自动分箱和手动优化步骤共产生了34个高质量的细菌MAGs(超过90%完成,少于5%冗余)和70多个中等质量的MAGs(超过50%完成,少于10%冗余)。展示了MEM对人类肠道活检的处理如何使从这些样本中重建MAG成为可能。对于34个高质量的MAG,我们计算了检测结果,它报告了给定参考序列中被给定宏基因组中至少一个短读序列的核苷酸的比例。因此,检测是一种非常有效的方法,能够讨论给定样本中给定种群的存在,独立于序列覆盖率,并通过避免由于非特异性序列招募而产生的假阳性。为了确认从MEM处理过的样本重建的MAG是未经处理的活检样品中真实存在的MAG,作者评估了比对回MAG时对照序列覆盖率的均匀性(图5c)。为了进行这一分析,作者选择了一种MAG,可以分辨出在对照样品中检测到最高的已知肠道微生物。对照样本显示,29个重叠群的读序分布均匀,表明该MAG也存在于对照样本中,但测序深度的限制阻碍了基因组的重建。总的来说,我们观察到,与对照样品相比,MEM处理样品中所有34种高质量MAG的检出率更高(图5d)。
为了量化来自对照样本的测序序列是否比对回了所有34个高质量MAG,本研究也绘制了每个MAG的检测图(图5d)。接下来,为了评估这些MAG是否受到污染,研究人员对每个基因组进行了分类。在95%的平均核苷酸一致性(ANI)阈值下,33个MAG被成功分类。研究人员也将每个分类MAG的大小与基因组分类数据库(GTDB)中匹配的参考基因组进行了比较,发现与当前微生物数据库高度一致(R2 = 0.78, P = 4.32 × 10−12),表明由MEM处理的样品构建的MAG不是人工产物(附表9和10)。一个梭杆菌MAG与已发表的粪便来源的MAG在86.85%的ANI上非常匹配,但GTDB无法分配物种水平的分类(附图7)。因为所有的MAG都是以相同的方式构建的,并且具有相似的质量指标,所以这个梭杆菌MAG很可能是一个未表征的分类单元,而不是污染。我们还想量化我们可以用MAG捕获的微生物多样性的范围。这34个MAG跨越了6个细菌门(图5d),从参与者CT12构建了一个古菌(Methanobrevibacter smithii))MAG,表明MEM处理的活组织检查能够重建古菌和各种细菌的基因组(附图4)。使用MEM,从人类肠道活组织检查的微生物中重建了高质量的微生物MAG,相对丰度低至1%。
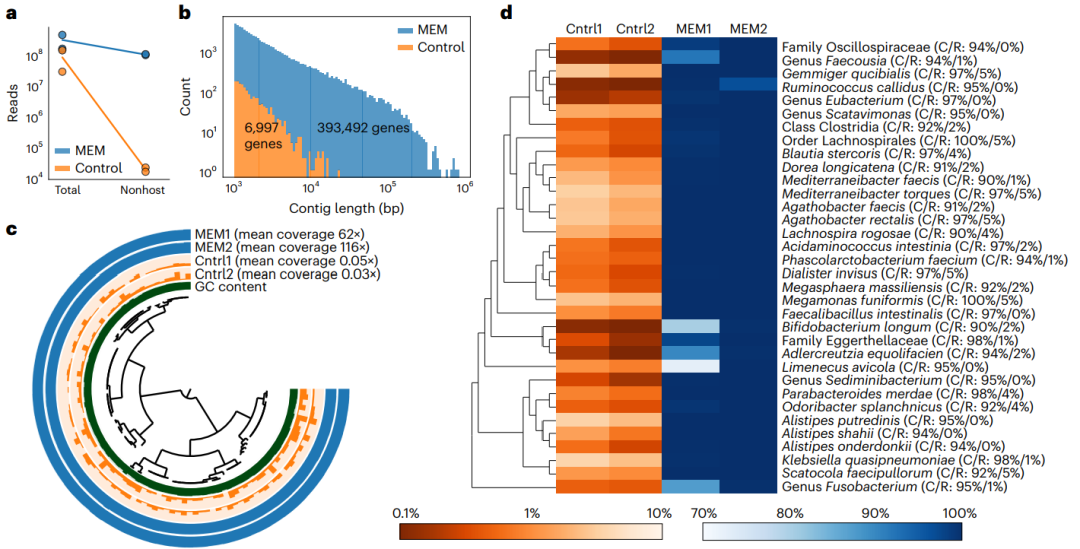
图5 通过宏基因组测序对经MEM处理的人肠道活检进行MAG构建。
a、对来自同一参与者(CT18)和肠道区域(升结肠)的两个对照和两个MEM处理的活检样品进行随机测序。在与人类参考基因组比对后确定非宿主读序的数量。
b、将每个条件下的两个样本聚类构建重叠群,并绘制重叠群长度的分布图。显示了在这些组合中鉴定的原核基因的数量。
c、Alistipes putredinis的MAG是由共同组装的MEM活检构建的。条形图的高度表示平均覆盖率,并对每个样本进行独立缩放。
d、从MEM活检的集合中,重新构建了34个高质量的MAG(超过90%完成,少于5%冗余)。热图显示了每个基因组至少被样本覆盖一次的百分比(即检测或呼吸覆盖),在对照样本中最大为3.7%,在MEM样本中最大为99.999%。在所有MAG中,MEM1、MEM2、Cntrl1和Cntrl2的平均检出率分别为97.3% (s.d 6.4%)、99.8% (s.d 0.7%)、1.2% (s.d 1.1%)和0.8% (s.d 0.7%)。对每个MAG进行分类,并在右侧列出完成度和冗余度(C/R)。系统发育树热图的左侧突出显示了每个MAG的分类分组。
MEM识别个体之间不同的微生物菌株
MEM identifies distinct microbial strains across individuals
在确定直接从人类肠道活检中构建MAG的可行性之后,我们接下来研究了微生物基因组在个体之间和个体内部的差异。为了确定MEM是否能够区分单个分类单元内个体之间微生物的群体水平差异,研究人员对来自参与者CT12的6份活组织标本进行了重新测序,测序深度约为2.5亿读序。分别对六个活检组织中的每一个进行组装和分组,并在样本中重复MAG。从参与者CT12构建并注释了在所有参与者中发现的最普遍和最丰富的Phocaeicola vulgatus物种的MAG (图6a)。然后,从所有五名参与者身上提取的60份肠道活组织检查的序列比对到该MAG上,以确定其他参与者的活检中缺少哪些基因。
如前所述,通过宏基因组序列对自然发生的P. vulgatus种群进行了基因水平上的解析分析,并揭示了一个大的核心基因组和个体间差异发生的基因(图6a)。来自参与者CT12胃肠道区域活检的基因似乎是保守的(CT12样本的平均基因检出率超过96%)。一些高检测基因仅在一个或两个参与者(CT12或CT12和另一个参与者)中发现,我们将其定义为独特基因。为了评估这些基因是否在功能上不同,我们用COG数据库对基因进行了注释,以确定同源基因。在CT12特有的287个基因中,其中100个基因被COG注释,并对应于广泛的功能(附图8)。在两个参与者(即CT12和另一个个体)特有的基因簇中,约30%被注释(附图8)。MEM处理可以深入了解具有相似健康状况的个体在肠道中占据相同空间位置的同一分类单元的功能不同的微生物种群。
在个体的胃肠道可以检测到SNVs
SNVs detectable across GI tract regions within an individual
最后,本研究研究了MEM处理是否可以通过增加覆盖深度,通过单核苷酸变异(SNVs)研究低生物量样品中的微生物种群遗传学。在这项分析中,该研究分析了来自参与者CT12的MAGs作为参考基因组,并将来自CT12的回肠末端和降结肠的成对末端读序比对到这些组装的基因组上。在所有6个样本(3个回肠末端和3个降结肠)中,6个MAG的平均覆盖率超过50倍,并被选中用于后续的SNV分析(附图9)。通过将每个样本的配对末端读序与参考序列(MAG)进行比较,生成SNV谱。我们通过为单个回肠末端活检的另外三个技术重复准备文库(图6b),研究了PCR错误是否可能导致我们数据中观察到一些SNVs,期望技术重复的SNV谱差异应该是最小的。通过观察在一个、两个或全部三个重复中发生的核苷酸变异,我们观察到Ruminococcus bromii与参考核苷酸的最小偏差为21%(图6b),仅允许选择SNVs,并将群体结构分析中PCR错误的影响降至最低。利用固定指数对这些数据进行分析表明,一些分类群,如R. bromii(图6c)和Gemmiger formicilis(附图9),是由上下肠道不同的亚群组成的。为了评估这些SNVs在功能上是否重要,我们对R. bromii的SNVs进行了密码子水平和翻译(氨基酸)分析,并根据位置检测了类似的活检聚类(扩展数据图5)。通过更深入的测序和活检样本中MAGs覆盖率的增加,SNVs的恢复使我们能够检测单个个体下胃肠道中某些个体分类群的亚群结构的存在。
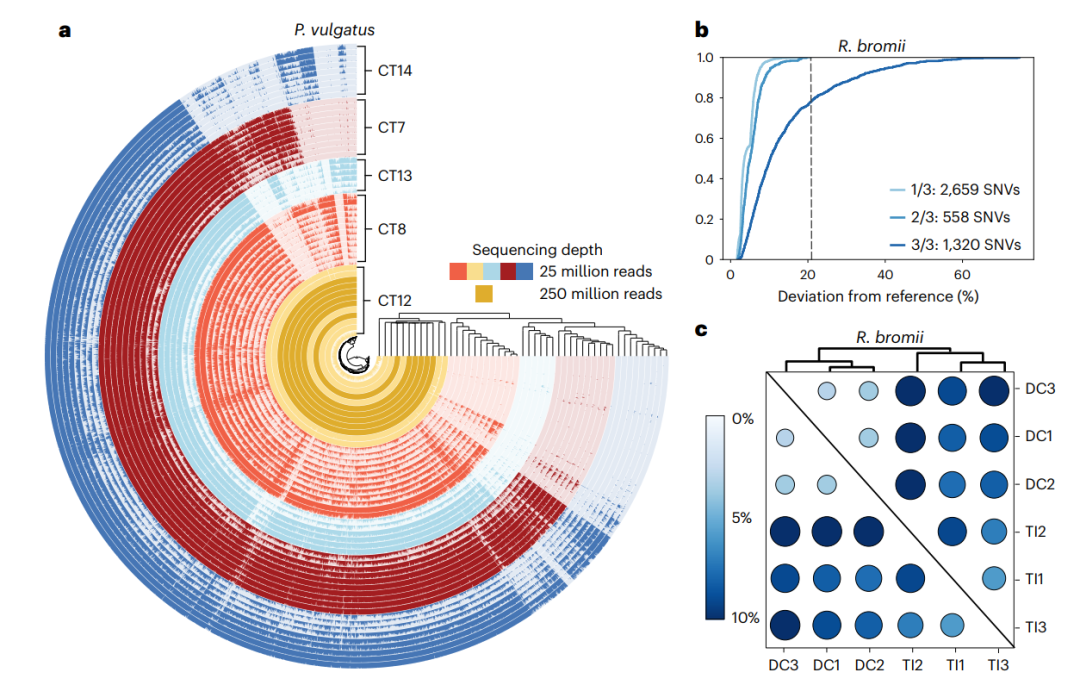
图6 沿胃肠道存在的个体间和个体内细菌多样性。
a,对所有5名参与者进行了P. vulgatus基因水平分析。通过基因检测对样本进行分组,定义为每个基因的覆盖率至少为1×coverage的百分比,并显示出强烈的参与者依赖性分组,但缺乏根据胃肠道位置进行分组。
b、R. bromii MAG中SNVs发生的经验累积分布函数以及这些SNVs在三个技术重复中与参考值的偏差。1/3、2/3和3/3表示在该位置具有SNV的技术重复数,然后是每个类别中SNV的总数。在与参考值偏离21%处绘制黑色虚线;高于此值,所有观察到的SNVs都存在于所有三个技术重复中。
c,对所有样品的平均覆盖率超过50倍的MAG进行核苷酸水平分析。图中显示的是在R. bromii的编码区域内分析的SNVs的固定指数,与参考集的最小偏差为21%。根据固定指数对样本进行聚类,可以看到很强的区域依赖性分组。DC:降结肠;TI,回肠末端。
- 讨论 -
MEM适用于富含哺乳动物宿主的样本,它使得对这些样本中存在的微生物进行宏基因组测序和分析成为了可能。MEM能够从固态哺乳动物组织中去除超过1000倍的宿主DNA,并同时最小化对微生物群落组成表征的干扰。MEM方法简单且快速,处理时间不到30分钟,便于集成到实验室或临床工作流程中,无需现场培训。MEM与微生物DNA的宏基因组测序高度兼容,与相似测序深度的对照样本相比,能检测到超过400倍的微生物种类和基因,同时也包括低丰度种类。MEM能够直接从人类肠道活检中以低至1%的相对丰度对整个微生物基因组进行培养独立组装。MAG的组装使研究个体之间和个体胃肠道内的亚群变化成为了可能。
本研究开发的MEM方法也有以下局限性。研究人员已分析过具有少至102个16S rRNA基因拷贝/µl的活检样本(相当于每毫克组织大约有104个16S拷贝),但要深入分析这一细菌载量以下的样本,需要更大程度的宿主去除率和/或更深的测序深度。所以研究人员也建议用户参考附图1,根据细菌载量预测非宿主读序的百分比,以指导测序的深度。本研究也已成功地将MEM应用于了健康的肠道活检样品中,但对于可能干扰分析的特殊样本,例如有活跃炎症或出血的样本,还需要进行额外的验证。对于保存的组织样本,也需要额外的验证。本研究仅研究了MEM对细菌和古菌的影响,未来的研究将揭示MEM是否会影响真菌组和病毒组。
本研究对鼠类粪便、肠道刮片、大鼠结肠部分、人类唾液和人类肠道活检验证了MEM。为了扩展MEM的使用范围,将来需要对植物、昆虫和其他非哺乳动物宿主的样本进行优化和验证。使用MEM进行样本处理还将实现更高的吞吐量和更低成本的微生物组调查,即使是在宿主含量适中的样本中。例如,在唾液中,从10%增加到95%的微生物序列可以将测序成本降低一个数量级。在临床研究中,我们预计MEM将为研究者研究肿瘤微生物组、黏膜肠道微生物组、肠道微生物的组织移位以及与复杂免疫疾病、免疫调节和癌症发展相关的组织相关微生物的作用提供帮助。
参考文献
Natalie J. Wu-Woods, Jacob T. Barlow, Florian Trigodet, Dustin G. Shaw, Anna E. Romano, Bana Jabri, A. Murat Eren & Rustem F. Ismagilov. (2023). Microbial-enrichment method enables high-throughput metagenomic characterization from host-rich samples. Nature Methods, https://doi.org/10.1038/s41592-023-02025-4
- 作者简介 -
通讯作者

加州理工学院
Rustem F. Ismagilov
教授
Ethel Wilson Bowles and Robert Bowles Professor of Chemistry and Chemical Engineering; Merkin Institute Professor; Director of the Jacobs Institute for Molecular Engineering for Medicine。专注于微生物与人类宿主在不同情况下的相互作用关系,从致病到有益微生物的定植,再到两者之间的一切互作关系。至今已在国际主流刊物Cell, Science, Nature, Nature Methods, Nucleic Acids Research, Microbiome等刊物发表学术论文80余篇,引用超过35000次,h-index 87。
主页:https://cce.caltech.edu/people/rustem-f-ismagilov
猜你喜欢
iMeta简介 高引文章 高颜值绘图imageGP 网络分析iNAP
iMeta网页工具 代谢组MetOrigin 美吉云乳酸化预测DeepKla
iMeta综述 肠菌菌群 植物菌群 口腔菌群 蛋白质结构预测
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature
一文读懂:宏基因组 寄生虫益处 进化树 必备技能:提问 搜索 Endnote
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流快速解决科研困难,我们建立了“宏基因组”讨论群,己有国内外6000+ 科研人员加入。请添加主编微信meta-genomics带你入群,务必备注“姓名-单位-研究方向-职称/年级”。高级职称请注明身份,另有海内外微生物PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。
点击阅读原文,跳转最新文章目录阅读


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









