






密集的深海热液垫层中病毒与跨越远缘微生物域的宿主相互作用
Viruses interact with hosts that span distantly related microbial domains in dense hydrothermal mats

翻译:周之超@UW-Madison
Article,2023-4-06,Nature Microbiology, [IF 30.96]
DOI:10.1038/s41564-023-01347-5
原文链接: https://www.nature.com/articles/s41564-023-01347-5
第一&通讯作者:Yunha Hwang
通讯作者: Peter Girguis
主要单位:
Department of Organismic and Evolutionary Biology, Harvard University, Cambridge, MA, USA
哈佛大学有机和进化生物学系
- 摘要 -
自然界中的许多微生物居住在密集的、新陈代谢相互依赖的群体中。我们通过研究生物量密集的深海热液垫层中微生物-病毒的相互作用,调查了微生物-病毒相互作用的性质和程度与微生物密度和互养的关系。通过使用宏基因组测序,我们发现在垫层中,有许多系统发育上相距甚远的(最高到域级)微生物对相同的病毒编码了基于CRISPR的免疫力。在已知的互养微生物伙伴之间病毒与宿主的相互作用跨越了微生物域的证据特别引人注目,例如那些从事厌氧产甲烷的微生物伙伴。这些模式被基于接近连接(Hi-C)的结果所证实。对公共数据集的调查显示,在已知有互养作用生物膜的各种生态系统中,有更多的病毒与宿主进行跨域互动。我们提出,病毒颗粒和/或DNA进入非主要宿主细胞可能是生物量密集的生态系统中的一种常见现象,对互养微生物和CRISPR介导的种群间抗病毒能力的增强具有生态进化意义。
- 引言 -
自然界中的大多数细菌和古菌都是以聚合体或生物膜的形式出现的。这些微生物聚集体通常由系统发育上相距甚远的生物体组成,从事相互依存的代谢活动(例如,h互养)。然而,大多数宿主与病毒的相互作用是在均匀的液体培养物中研究的,我们对密集的、基质结合的和异质的生物膜中宿主与病毒的相互作用的理解还有许多差距。特别是,在复杂的微生物群落中,基因不同和系统发育遥远的微生物在高度接近的情况下共存,并进行高度嵌套的新陈代谢,在宿主范围、病毒生命周期、传播方式和宿主-病毒共同进化等方面存在重大问题。
一般来说,人们认为病毒感染的宿主范围很窄。然而,最近的研究表明,广泛的宿主范围的病毒可能在自然界中更常见,可能由于培养的偏见而被忽视。到目前为止,已有关于病毒感染多个细菌种类、目,甚至可能是门的报道。此外,病毒的宿主范围也被证明是一个动态特征。值得注意的是,最近的一项研究报告说,噬菌体的吸附和进入细胞并不等于完全完成裂解周期,这表明病毒可能与更多样化的细胞互动,在这些细胞中可以进行完整的感染周期。
我们假设,更广泛的宿主范围的病毒可能在以互养代谢为主的生物膜中普遍存在,这是因为与系统发育不同的微生物的接触时间延长,以及由胞外聚合物质(EPS)和空间异质性引起的有限的宿主和病毒散布和/或生境范围。为了解决这一假设,我们对深海热液微生物垫层中的病毒基因组和任何病毒与细菌或古菌的相互作用(以下称为宿主-病毒的相互作用)进行了描述,这些垫层是热液喷口周围无处不在的化合自养生物膜。这些垫层由非常密集的、新陈代谢耦合的细菌和古菌群落组成,在温度和地球化学方面具有鲜明的空间梯度和时间变化。我们表明,在系统发育上相距甚远的(即来自不同门甚至域的分类群)、具有互养代谢能力的微生物,往往对垫层中的相同病毒进行基于CRISPR的免疫。这种模式没有从物理上相邻的热液羽流样品中检测出来,这些样品的生物量较低,代谢相似的群体。此外,根据Hi-C接近连接测序,这些微生物基因组与相同的病毒基因组表现出共同定位的特点。通过检查公开的宏基因组,我们还发现病毒与其他已知含有互养生物膜的生态系统中的细菌和古生物分类群相互作用。我们通过检查病毒基因组中的辅助代谢基因(AMGs),以及识别经历选择的病毒和微生物基因,进一步研究了这些宿主-病毒互动的生态进化意义。最后,我们提出了病毒与互养宿主多价互动的四个模型,并讨论了它们对微生物进化的影响,特别是在水平基因转移、基因多样化和CRISPR介导的全群落免疫记忆方面。
- 结果 -
互养和代谢性相互依赖的微生物垫层
RR2107考察于2021年11月11日至12月5日在墨西哥的Guaymas盆地进行。在遥控潜水器(ROV)Jason的J2-1398次潜水中,从一个连续的热液垫层中回收了10个pushcore(直径7.5厘米,长30厘米)。该垫层在温度和化学性质上都是不均匀的,地下温度在21℃和53℃之间(图1a,b)。从表层垫层和顶层沉积物中提取的DNA被用作宏基因组测序的模板,产生了18亿个150bp读数对。我们利用基于读数覆盖率、k-mer频率和/或接近连接数据的基因组分档,从整个垫层中恢复了303个中、高质量的代表性微生物宏基因组组装基因组(rep_mMAGs)(方法)。在10个样本中,8个样本由5个遗传上确定的(平均核苷酸相似度(ANI)97%,物种水平)的Gammaproteobacteria种群主导,都属于Beggiatoaceae家族(图1c),具有硝酸盐耦合硫氧化的基因组能力(补充表3)。另外两个样品(M2和M7)显示出较高的物种均匀性(香农多样性指数)。超过一半(n = 153)的种群在单个样品中被唯一检测到,只有3个rep_mMAGs(Gammaproteobacteria_19_1、Campylobacteria_146_1和Acidimicrobiia_30_1)在所有10个样品中被检测到。尽管垫层在形态和环境上有明显的高度斑驳性,但在丰富的硫氧化细菌(SOB)种群组成变化的驱动下,垫层的微生物群落组成可以归纳为两个空间上有组织的集合(扩展数据图1b),这表明物理上的接近可能在群落组装中起到更大的作用。环境条件(例如温度或硫化氢)的高变异性可能是较罕见的种群的样本特定变化的原因,这可以通过测量这些生物能够代谢的其他地球化学物质(例如甲烷和氨)来进一步探索。如前所述,这些微生物垫层以化合自养细菌和古菌为主,主要靠地热衍生的还原硫、氮和碳水化合物维持,303个rep_mMAGs中分别有223、192和40个编码了至少一个参与硫、氮和甲烷代谢的基因。这些热液沉积物中的微生物代谢被认为是高度相互依存的,以前的研究发现了互养厌氧甲烷氧化(ANME)古菌和硫酸盐还原菌(SRB)的证据,将厌氧甲烷氧化与硫酸盐还原结合起来,以及假设的SRB和SOB之间基于硫的互养作用。值得注意的是,这些rep_mMAGs中有76%编码了氢化酶、c型细胞色素和/或PilA,表明底物介导的和/或直接的种间电子转移具有广泛的可能性。
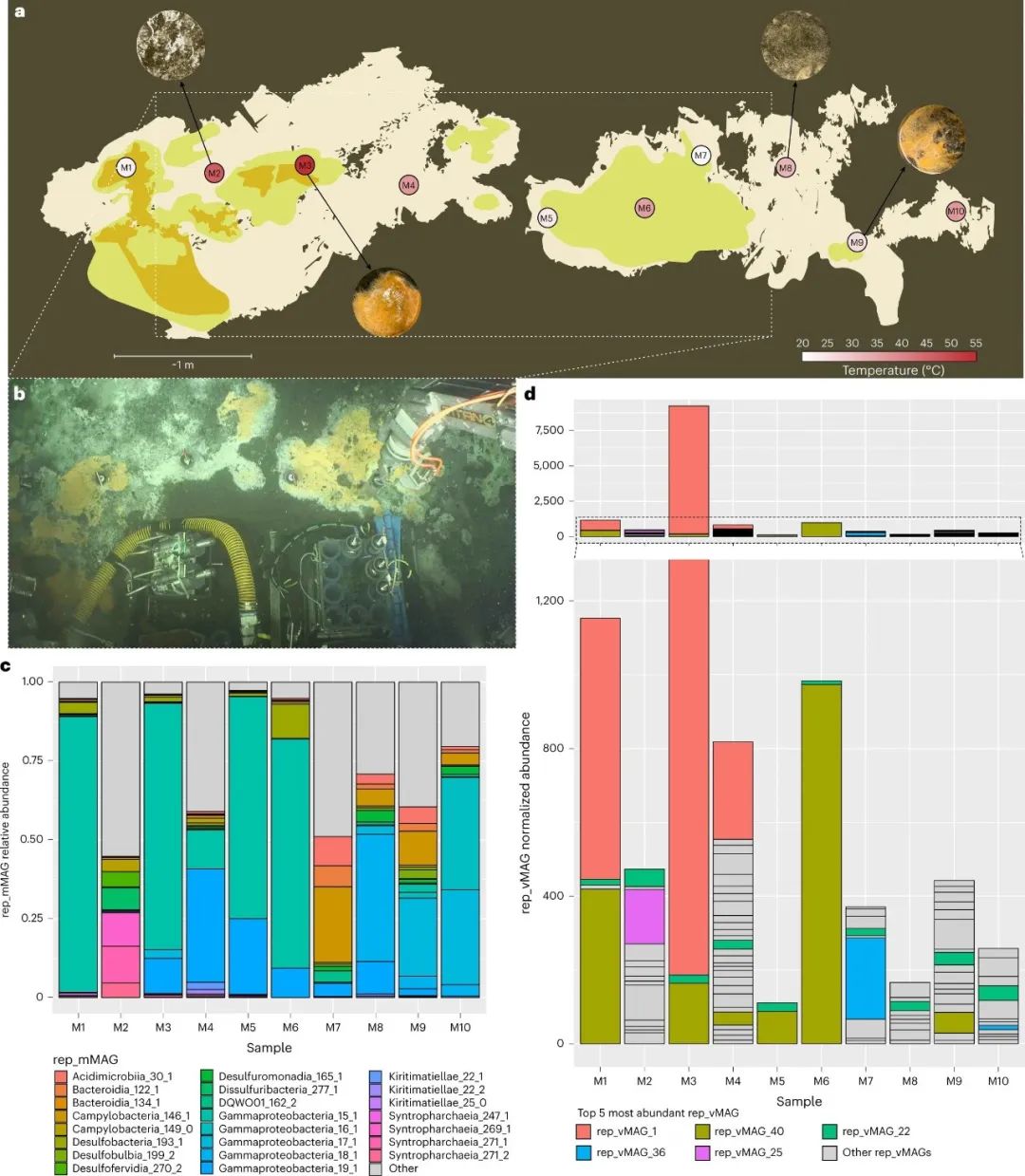
图1:高度异质但连续的深海热液垫c层
a, 采样的微生物垫层的视觉示意图。采样位置是根据采样期间观察到的三种主要颜色(橙色、黄色和白色)来说明的。距离和形状是近似的,是利用ROV Jason潜水期间拍摄的高分辨率视频和照片重建的。Pushcore的位置是根据现场的温度来标示的。
b, 取样垫层中间部分的俯视图(大约在a中被勾勒成虚线框)。
c, 每个样品中前10个最丰富的rep_mMAG的相对丰度(物种水平,97% ANI cutoff)。
d,每个样品中47个高质量或完整的rep_vMAGs(95%ANI cutoff)的归一化丰度。前5个最丰富的rep_vMAGs用颜色表示。
近乎完整的病毒基因组的特征分析
在10个宏基因组中,我们恢复了47个有代表性的病毒MAGs(rep_vMAGs,在95%的ANI中去复制),根据CheckV使用高置信度的完整性估计方法(详见方法),这些MAGs是完整的(n = 27)或高质量的(n = 20)。这些rep_vMAGs的大小差异很大,从12kbp到437kbp不等。只有两个rep_vMAGs(vMAG_46和vMAG_25)用CheckV检测为原病毒。rep_vMAGs的丰度分布比rep_mMAGs的丰度分布更加异质,并表现出较少的基于邻近性的聚类。与微生物种群类似,一半以上(n = 26)的rep_vMAGs只在一个样品中被检测到(意味着栖息地范围小),而只有一个rep_vMAG(rep_vMAG_22)可以在所有10个样品中被检测。一个样品(M3)显示出大于7倍丰度的裂解病毒群体(rep_vMAG_1),这与最近的病毒感染相一致(图1d)。病毒多样性与微生物物种多样性高度相关(皮尔逊相关系数:0.83,n = 10,P = 0.00293),尽管没有发现微生物和病毒组成之间的统计学意义上的共同对应关系(sCoCA,n = 10,P > 0.05)。相对于参考病毒基因组所占据的遗传多样性空间,本研究中回收的rep_vMAGs表现出非常高的分类学和基因含量多样性。只有4个rep_vMAGs可以与参考病毒基因组在 "属 "的水平上聚类,并可以被归类为两个(以前指定的)Podoviridae,一个Myoviridae和一个Siphoviridae。值得注意的是,一个由7个rep_vMAGs组成的分类群与Flavobacterium噬菌体有遥远的联系,其中3个rep_vMAGs形成了一个新的属级群,与任何特征化的参考病毒序列没有相似的基因。大多数(49个中的29个)与参考病毒基因组或彼此之间的基因内容没有高度相似性。许多病毒基因组含有新的辅助代谢基因(AMG),如含Rubisco大结构域的蛋白、含醛缩酶II结构域的蛋白、含硝基还原酶结构域的蛋白、磷酸盐饥饿诱导蛋白PhoH和抗蒺藜蛋白TerD。我们还检测到宿主-病毒军备竞赛的证据,一些病毒基因组编码防御机制,如RelE/StbE家族毒素、HigA家族解毒剂和一个假定的中止感染蛋白。
微生物CRISPR-Cas位点
本研究中回收的rep_mMAGs具有多样化和丰富的CRISPR-Cas系统。我们在303个rep_mMAGs中的119个检测到317个cas位点。一个群体基因组中的cas位点数量在1到16之间,其中一个ANME-1 rep_mMAG(Syntropharchaeia_272_1)编码了16个cas位点,属于不同的亚型(6个1类IB亚型,3个1类IIIA亚型,2个1类IIIC亚型,1个1类I型,2个1类III型,以及2个未分类的集群)。此外,我们在65个遗传学定义的群体中发现了116个独特的CRISPR重复序列。这些CRISPR重复序列通过序列相似性(>95%的核苷酸相似度(ID))被聚类为102个聚类。大多数(91%)检测到的CRISPRs是特定于一个种群的,80%的CRISPR编码种群与最多2个独特的CRISPRs有关。然而,我们观察到相同或接近相同(>95% ID)的CRISPR重复序列在系统发育遥远的种群中共享。这些CRISPR位点有可能是水平转移的,但我们不能排除因其重复性和差异性导致的分选错误的可能性。由于将一个特定的宿主分类群分配给一个重复序列的模糊性,这种跨分类群检测到的CRISPR重复序列被排除在基于间隔的宿主-病毒匹配之外。此外,我们发现种群(Gammaproteobacteria_17_1、Desulfobacteria_193_1、Desulfobacteria_189_1)编码多达6个不同的CRISPR重复序列,可能代表CRISPR位点的种群内多样性。没有发现独特的CRISPRs的数量与rep_mAG的大小、相对丰度或栖息地范围之间的相关性。
重建历史上宿主与病毒的相互作用
利用群体特异性CRISPR重复序列,我们在10个宏基因组中挖掘了278,929个独特的间隔体。间隔体-to-原间隔体(病毒基因组中作为间隔体模板的区域,随后被CRISPR-Cas系统锁定)在rep_mMAGs和rep_vMAGs之间的匹配被用来推断宿主对特定病毒的适应性免疫,从而推断历史上宿主-病毒的相互作用。我们确定了28个rep_vMAGs和29个rep_mMAGs之间的96个相互作用,这些相互作用来自与39个rep_mMAGs特异性CRISPRs相关的22,466个间隔体-to-原间隔体匹配。一小部分(0.01%,25个间隔体)的原间隔体被发现在最多2个rep_vMAGs的非独特区域。大多数(66%)CRISPRs与病毒目标缺乏高置信度的匹配,这表明病毒种群可能存在比用宏基因组测序所能检测到的更高的多样性,和/或在这种环境中病毒种群的快速周转。在图2a中,我们显示了基于CRISPR-间隔体的宿主-病毒相互作用,宿主-病毒对至少有两个不同的间隔体-to-原间隔体匹配。大部分(92%)的间隔体到原间隔体的匹配是在rep_vMAG_1和3个Gammaproteobacterial rep_mMAGs之间,这与rep_vMAG_1的观察到的归一化丰度是一致的,它比其他rep_vMAGs高几个数量级(图1d)。我们观察到一个惊人的模式:已知的和假设的互养伙伴(ANME-SRB,SOB-SRB)基于CRISPR-间隔体匹配相同的rep_vMAGs。图2a中可视化的宿主-病毒匹配是使用独特的CRISPRs进行的,代表了可能来自历史上的相互作用的免疫,而不是CRISPR阵列的横向转移。此外,来自不同微生物种群的CRISPR-间隔体匹配分布在整个病毒序列中,在针对特定的基因组区域方面没有表现出偏好(例如,图2b)。有趣的是,我们发现rep_vMAG的大小与它们可以使用CRISPR间隔体找到的宿主数量之间存在统计学上显著的正相关(Pearson双侧相关系数=0.8,调整后P<1×10-6,n=36),表明分类学上不同的微生物的CRISPR靶向锁定可能对较大的病毒更常见。
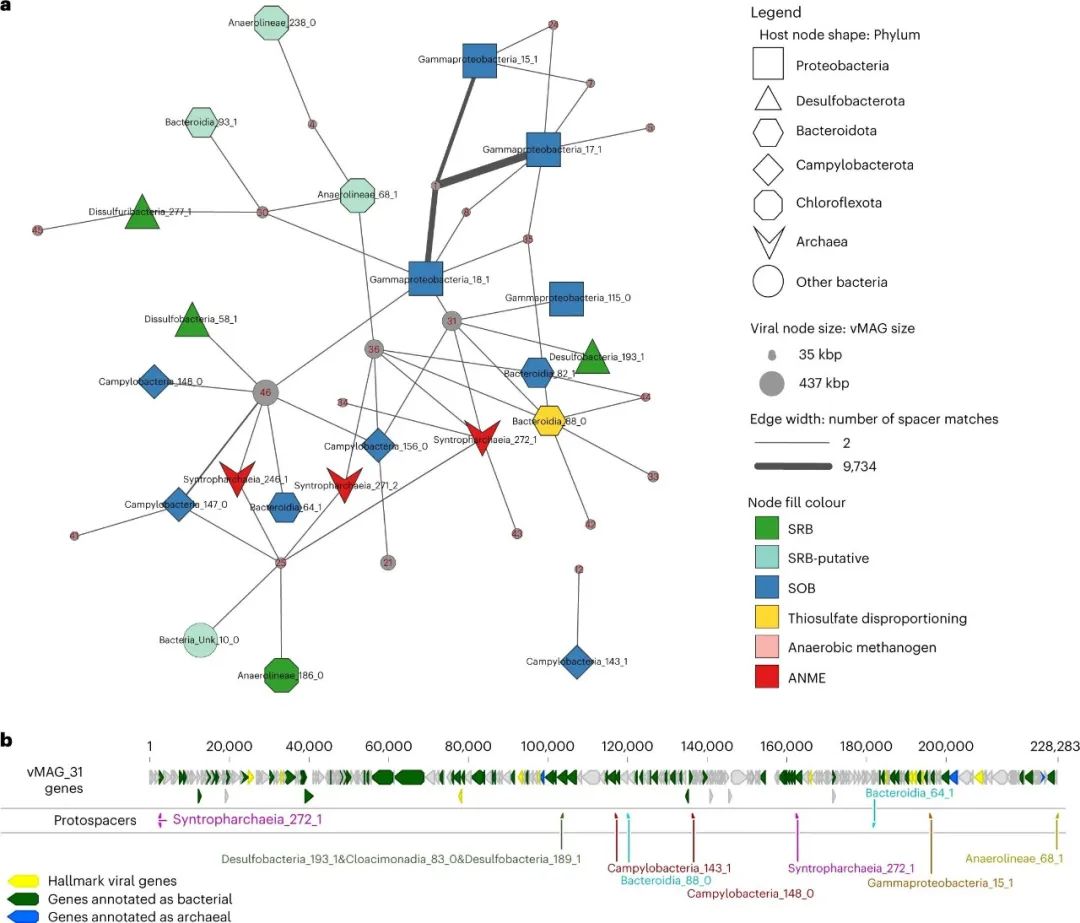
图2:根据CRISPR 间隔体-to-原间隔体的匹配,展示修剪过的历史上的宿主-病毒相互作用。
a, rep_mMAGs和rep_vMAGs之间的间隔体-to-原间隔体匹配,其中至少发现两个不同的匹配,用一条边表示。该网络中排除了在多个rep_mMAG中发现的CRISPR重复序列。边缘的宽度对应于不同匹配的数量。宿主节点的形状和颜色分别表示宿主系统和推测的代谢。病毒节点的大小与相应的rep_vMAG长度成比例。
b, 沿着病毒contig的原间隔体匹配的可视化,其与属于不同门和域的至少八个宿主特有的CRISPRs相关。
Hi-C接近连接显示宿主-病毒基因组的联系
虽然CRISPR间隔体到原间隔体的匹配提供了关于宿主和病毒之间历史性相互作用的高置信度信息,但一些宿主并不编码CRISPRs,而且CRISPR阵列由于其重复性,往往不能在shotgun组合中组装。原位宿主-病毒基因组的联系可以用接近连接法进行探测。我们构建并测序了10个Hi-C宏基因组文库(共15亿个Hi-C 150-bp reads对),这些文库编码了推定的染色体接触信息,包括细胞内病毒和宿主基因组之间的接触。我们使用分档的contigs(原始噪信比=0.021±0.016)和分档的contigs的目水平分类(relaxed噪信比=0.016±0.011;噪信比的计算见方法)来估计Hi-C接触的噪信比。我们检测到病毒和宿主contigs之间有5292个联系,这些联系可合并为属于31个不同门的36个rep_vMAGs和241个rep_mMAGs之间的859个联系(其中24%在多个样本中被复制),揭示了宿主和病毒之间潜在相互作用的高度嵌套网络(图3)。经过归一化处理,我们观察到一些宿主-病毒基因组的相互作用在宿主和病毒contigs之间的独特联系数(图3中可视化为边缘宽度)和宿主-病毒对之间的最大联系强度(一对病毒和微生物contigs之间的最大归一化接触数;可视化为边缘的暗度)方面都更加明显。这些更明显的宿主-病毒联系可以解释为宿主-病毒对之间存在原位感染的信号,其中Hi-C reads可以捕获在某些宿主细胞内积极复制的病毒基因组。在某些情况下,我们可以将样品M3中rep_vMAG_1和Gammaproteobacteria_15_1之间的强接近连接信号与vMAG和mMAG覆盖率的增加和该样品中rep_vMAG相对丰度的数量级增加(图1d)联系起来。然而,需要注意的是,对于大多数其他的宿主-病毒对来说,这些显著的Hi-C连接的测量结果并不一定与更高的群体水平感染率相关,因为在交联时对感染事件的Hi-C捕获是相对罕见的。在这里,我们利用这些信号来确定病毒的潜在主要宿主,并分解其多价互动。例如,我们观察到在不同的样品中,病毒与多个宿主相互作用的模式是一致的,但与一个子集的宿主的相互作用更为显著,而不考虑样品之间的病毒和微生物丰度的高度变化。我们观察到Hi-C连接与一些宿主-病毒群体中的CRISPR-间隔体匹配相重叠(可视化为红色边缘),可能反映了基于CRISPR的免疫中的群体内和/或菌株级的异质性(其中宿主群体的不同亚群/菌株拥有针对不同病毒的CRISPR免疫)。我们基于Hi-C连接的宿主-病毒相互作用网络与我们使用基于CRISPR的方法所观察到的一致,病毒与具有相互依赖的新陈代谢的系统发育遥远的生物体共同定位。宏基因组中的Hi-C连接包含固有的噪声,因此我们不能排除一些推断的宿主-病毒连接可能是假阳性的可能性。然而,CRISPR和邻近连接数据之间的一致结果表明,病毒-微生物相互作用网络在这个热液垫中的嵌套程度比通常观察到的或预期的要高。我们观察到一种模式,即较大的rep_vMAGs(病毒节点大小)与系统发育不同的rep_mMAGs表现出更多的(更多的边缘)和显著的(更厚和更深的边缘)联系,与CRISPR数据中观察到的模式相似。然而,在Hi-C reads信号上存在明显的对contig长度和覆盖率的偏向,即使在归一化后也不能完全控制;因此,这一观察需要使用对contig长度偏向较小的补充方法(例如,单细胞病毒标记)的进一步检查。有趣的是,我们发现rep_vMAGs的核苷酸多样性、平均丰度或栖息地范围与基于CRISPR和基于Hi-C方法推断的宿主范围之间没有相关性。
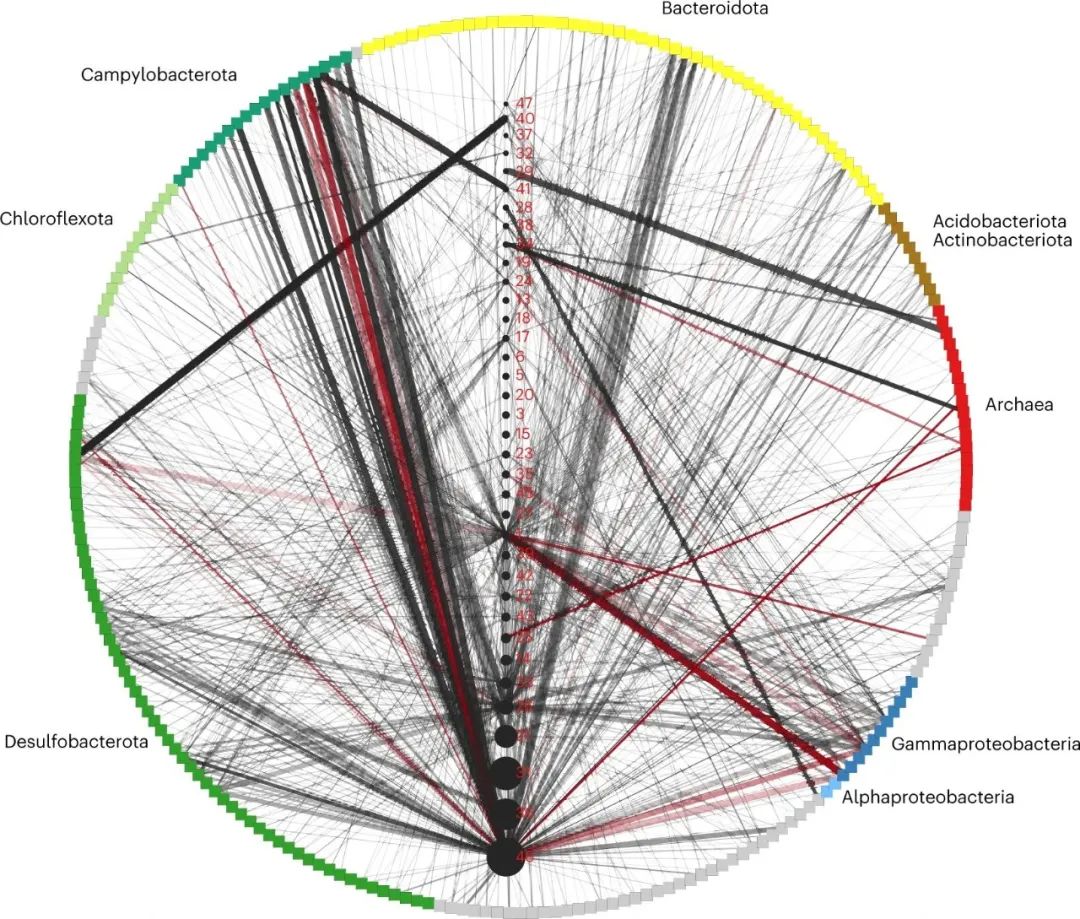
图3:Hi-C接近连接反应原位宿主-病毒相互作用网络。
rep_mMAGs和rep_vMAGs基于归一化的Hi-C接触的网络可视化。rep_mMAGs被定位在一个圆形的方形节点中,颜色代表分类学分类(灰色:其他)。rep_vMAGs被垂直定位在沿着中心的黑色圆形节点中,rep_vMAG大小不断增加。rep_vMAG的ID用红色标签表示(例如,1指rep_vMAG_1)。边缘的厚度代表contig-to-contig链接的数量,而边缘的暗度与一个宿主-病毒对中任何两个contigs之间Hi-C接触的最大归一化强度相关。以前用CRISPR-间隔体匹配检测到的宿主-病毒对用红色表示。
与热液羽流水样的比较
我们假设,微生物垫层的密度和空间结构有助于基于CRISPR的免疫网络的嵌套模式。为了探索这种关系,我们对10个受热液影响的水样(以下简称热液水(HW)样本)的宏基因组进行了同样的基于CRISPR的宿主-病毒网络分析,其中9个样本来自附近的热液羽流(距垫层约45公里),1个样本来自取样垫层上的水。HW宏基因组与垫层宏基因组在测序深度和组合大小上都很相似(Welch's t-test,双侧,n = 20,P > 0.05)。我们对10个HW组合中的168个中-高质量的rep_mMAGs进行了分类,尽管在分类学上与从垫层组合中回收的rep_mMAGs不同,但这两个数据集具有相似的代谢能力和相似的物种均匀性水平(Welch's t-test,双侧,n = 20,P > 0.05)。尽管HW样品之间的物理距离较大,但HW样品的微生物群落比垫层样品更均匀。与垫层样品类似,HW样品由两种硫氧化型Gammaproteobacteria(HW_Gammaproteobacteria_164_1, HW_Gammaproteobacteria_163_1)主导。有趣的是,我们观察到在HW组合中发现CRISPR位点的频率要比垫层组合低一个数量级(Welch's t-test,双侧,n = 20,P = 0.001)。此外,HW组合中只有12个CRISPRs可以与中等质量的MAGs相关联,导致基于CRISPR的免疫网络更加稀疏和不健全,只有一个SOB(HW_Gammaproteobacterira_162_1)和一个病毒之间有可信的相互作用。羽流和垫层样品之间的相似性,如地理上的接近性、群落代谢能力和测序深度,为比较提供了合理性和机会。羽流样本中CRISPRs的丰度较低,表明羽流群体对基于CRISPR的适应性免疫的依赖性较低。基于CRISPR的免疫的可转移性和特异性赋予了这一观察的生态意义,提出了在不同环境中如何选择这种免疫学记忆的问题。虽然这种比较揭示了垫层和羽流之间宿主-病毒相互作用的性质和程度的关键差异,但为了进一步解释,还有一些注意事项需要考虑:首先,羽流中基于CRISPR的免疫网络的稀疏性可能部分是由于回收的病毒片段的丰度和多样性较低,其中只有感染微生物和/或附着在大于0.22微米的颗粒上的那部分病毒被回收。其次,基于CRISPR的免疫力的差异不一定反映原位宿主和病毒相互作用的深层网络的模式。
微生物域交叉病毒的全球分布
为了描述宿主与病毒之间跨系统发育距离的普遍性,我们在公共数据库中寻找映射到古菌和细菌CRISPR位点中发现的间隔体的病毒。我们在来自5个地点的25个样本中检测到26种病毒(表1)。这些病毒是在有证据证明生物膜形成的生态系统中发现的(例如,厌氧消化器污泥、尾矿池中的石化废水和富含二氧化碳的地下含水层),并且使用各种方法(如共同发生网络、转录组和脂质组)强调了代谢的相互依赖性。此外,许多匹配的CRISPR间隔体是在已知的或假设的互养类群中发现的,如Methanosarcinales、Methanoculleus、Smithella和Thermotogales。如上所述,基于CRISPR的宿主-病毒相互作用推断仅限于丰富的CRISPR位点可以被组装和分装的环境。因此,这些跨越大系统发育距离的宿主-病毒相互作用有可能在自然界中比用这种方法能检测到的更常见、更普遍。例如,我们在大型宏基因组数据集的MAG中检测到很少被分档的高置信度(方法)的CRISPR位点,这些数据集缺乏微生物垫层,并且具有较低的微生物密度(以及可能较少的代谢互养),例如来自夏威夷海洋时间序列和百慕大大西洋时间序列的寡营养水样本,以及在旧金山海岸收集的典型的深海沉积物样本。
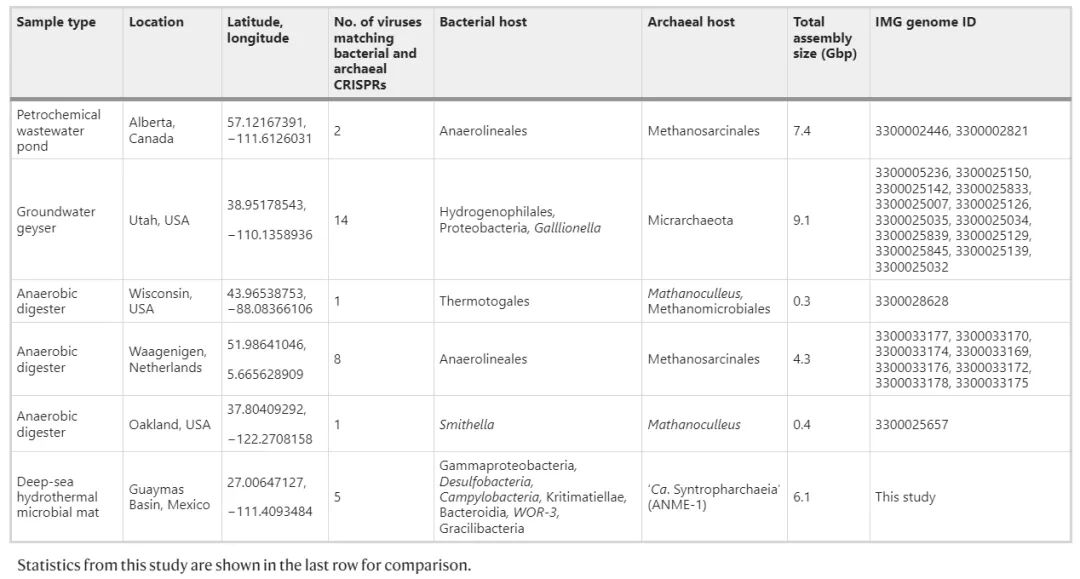
表1 公开的宏基因组中病毒与细菌和古菌的CRISPR间隔体相匹配结果
微生物垫层基因的选择和多样化
基于宿主-病毒相互作用网络的高度嵌套性和垫层样本之间病毒群落的高度异质性,我们假设病毒和微生物中经历选择的许多基因将分别与宿主范围和病毒防御有关。我们计算了病毒和微生物基因的pN/PS比率(非同义多态性与同义多态性的比率),并试图预测其功能。我们发现有18个病毒基因正在经历多样化的选择(pN/pS>2.5);然而,大多数基因不能被注释为具有某种功能。有趣的是,在4个被注释的基因中,有3个参与了DNA和RNA的代谢,如编码DNA导向的RNA聚合酶(RNAP)β和β prime(rep_vMAG_21)、DNA连接酶(rep_vMAG_31)和Superfamily II DNA/RNA螺旋酶(rep_vMAG_6)。我们还检测到一个含LamG结构域的蛋白(vMAG_4),可能参与了信号和细胞粘附,正在经历多样化的选择。rep_vMAG_21中编码RNAP的基因(RNAP1)具有最高的pN/PS比率,即4.9,有8个非同义突变散布在整个蛋白质中。值得注意的是,rep_vMAG_21有第二个编码β亚基的RNAP基因片段(RNAP2),它与RNAP1没有同源性,似乎也没有经历选择,可能有助于放松对RNAP1的纯化选择。RNAP1与以前表征的RNAP序列高度分歧,并扎根于Caudoviricetes多聚体RNAP支系的底部38。这个对RNAP1进行多样化选择的例子表明,这些病毒可能在加速通常在细胞生物中经历净化选择的housekeeping proteins的进化方面发挥了重要作用。经历多样化选择的微生物基因(pN/pS>2)包括编码参与各种防御系统的产物的基因,如II型毒素-抗毒素系统RelE/ParE毒素、HindIII家族II型限制性内切酶、III-B型CRISPR模块RAMP蛋白Cmr1,以及参与最近定性的PARIS和Septu抗噬菌剂库的基因。
- 讨论 -
在这项研究中,我们调查了微生物密度和代谢的相互依赖性如何在微生物垫层中形成宿主-病毒的相互作用。我们的研究结果综合起来提供了令人信服的证据,即在具有高生物量密度、多样性和代谢合作特点的微生物垫层和生物膜中,病毒可能与系统发育遥远的微生物相互作用。我们提出了四个非相互排斥的模型,以更好地理解这一意外观察的背景并提供潜在解释(图4)。在第一个模型中,我们提出病毒基因组可能进入病毒无法感染的细胞(即 "非主要宿主"),在这些生态系统中,病毒DNA和颗粒高度接近密集的、多样化的群落,这些群落的维持部分是由于微生物互养作用和EPS基质。第二种模式提出了病毒颗粒和/或基因组在互养微生物之间,甚至在不同域的微生物之间进行接触性转移的可能性。跨越大的系统发育距离的接合转移已被证实,并被假设为在自然界中更常见,我们的数据支持这一推测。在这两种情况下,将病毒基因组引入非主要宿主细胞会触发基于CRISPR的免疫反应,并导致间隔体的获得。这种机制可能导致整个种群对噬菌体的免疫记忆和反应增加,因此,当一个生物体的适应性与它的互养伙伴对噬菌体的抵抗力密切相关时,这种机制可能会被特别选择。这扩大了种群内泛免疫性的概念,可能包括跨种群和大系统发育距离的共享免疫性。特别是,这突出了微生物之间的代谢共生和 "防御性共生 "之间未被充分探索的联系。在第三和第四个模型中,我们提出了这样一种可能性:高密度的生态系统,如微生物垫层是病毒宿主转换和/或宿主范围扩大的热点。然而,单个噬菌体的这种变化和/或宿主范围的扩大仍有待实验证实(即通过病毒体生产的证据)。
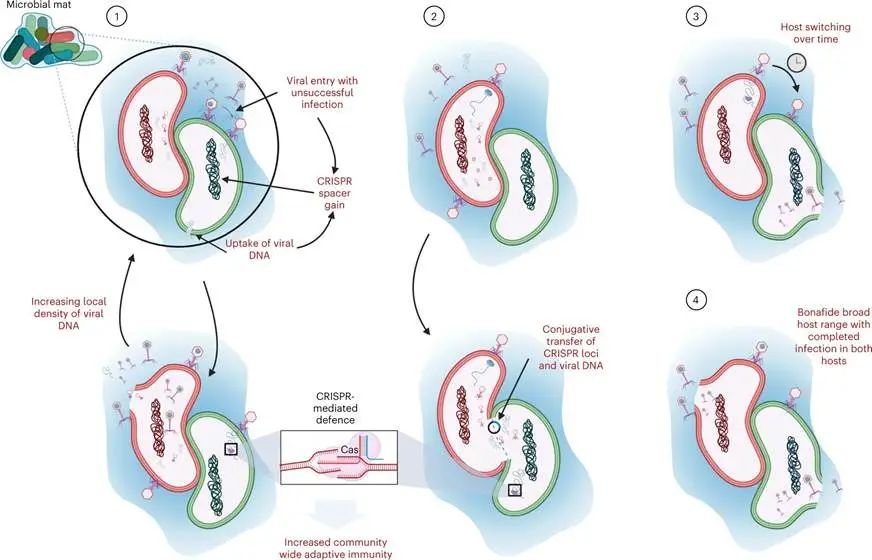
图4:在高微生物密度和代谢相互依存的生态系统中,宿主与病毒相互作用的四个可能模型。
红色和绿色的细胞代表系统发育上相距甚远且代谢独立的宿主(例如ANME-SRB)。蓝色阴影代表一个限制病毒和细胞外DNA扩散的EPS基质。在第一个模型中,我们说明了 "杂交 "病毒吸附和进入非主要宿主细胞(绿色)的可能性,这导致了CRISPR-间隔体的获得事件。另外,由于EPS的有限扩散潜力,在主要宿主的裂解事件后,可能导致病毒颗粒和病毒DNA的局部密度增加(红色)。因此,这可能导致非主要宿主细胞自然摄取病毒DNA的可能性增加,也会导致间隔体获得事件。在第二个模型中,我们提出了CRISPR阵列和病毒DNA的接触性转移的可能性。这也会导致非主要宿主细胞(绿色)获得一个CRISPR-间隔体事件。在这两个模型中,这导致了CRISPR介导的全群落范围内免疫记忆和抵抗作用的增强。在第三个模型中,我们提出了病毒宿主随时间转换的可能性,从T=0时的主要宿主(红色)到其最近的互养伙伴(绿色),因为初始宿主对病毒进行了进化。最后,在最后一个模型中,我们考虑了真实的广泛的宿主范围,在两个宿主中都成功感染病毒的可能性。
病毒和非主要宿主之间的这些相互作用具有广泛的生态和进化意义,包括但不限于:(1) 调解跨域和跨门的水平基因转移,(2) 在具有高微生物密度和互养的生态系统中增加适应性免疫的选择优势,(3) 参与防御和宿主范围扩展的微生物和病毒基因的多样化。我们的发现提醒人们不要仅仅依靠基于CRISPR的方法来推断病毒对宿主的感染性,因为非感染性的相互作用也可能导致CRISPR-间隔体的获得事件。我们的发现还强调了使用CRISPR-间隔体信息探索感染以外的宿主-病毒相互作用的潜力,如解析跨越大系统发育距离的水平基因转移网络和描述整个群落协作免疫的特征。此外,我们提出,这种嵌套的 "免疫网络 "可以用来产生关于新的微生物相互作用的假设(例如,互养作用)。从根本上说,在我们阐明高密度生态系统中共存、竞争和合作的基本机制时,这些扩大的宿主-病毒相互作用模型提出了一个重要的考虑层面。
参考文献
Hwang, Y., Roux, S., Coclet, C. et al. Viruses interact with hosts that span distantly related microbial domains in dense hydrothermal mats. Nat Microbiol (2023). https://doi.org/10.1038/s41564-023-01347-5
- 作者简介 -
通讯作者

哈佛大学
Peter Girguis
教授
通讯作者Peter Girguis于2005年加入哈佛大学,担任助理教授,并于2012年成为正式教授。他的研究工作旨在更好地了解微生物如何在地球生物圈中介导物质和能量流动。他为研究微生物介导的能量流动和采集开发了新的方法和技术,包括能更好地模拟环境条件的实验室和现场培养器,以及可现场部署的仪器,如水下质谱仪、碳同位素分析仪和高性能氢传感器,使他能够在实验室和现场研究微生物过程。Girguis撰写或合作撰写了超过85篇出版物,包括在Nature, Science, Proceedings of the National Academy of Sciences, Proceedings of the Royal Society上发表的论文。Girguis是海洋探索信托基金(OET)的董事会成员,是施密特海洋研究所海洋运载工具咨询委员会的成员,并曾担任国家科学基金会深潜科学委员会(DeSSC)的主席。Girguis的荣誉包括连续5年的杰出教学表彰,2007年和2011年林德伯格基金会科学与可持续发展奖,2010年ENI国际能源与环境奖的荣誉奖,2008年发现杂志的 "10个可以改变世界的日常技术"(生物动力灯)的专题,以及2008年巴克明斯特-富勒科学创新奖的荣誉奖。
猜你喜欢
iMeta简介 高引文章 高颜值绘图imageGP 网络分析iNAP
iMeta网页工具 代谢组MetOrigin 美吉云乳酸化预测DeepKla
iMeta综述 肠菌菌群 植物菌群 口腔菌群 蛋白质结构预测
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature
一文读懂:宏基因组 寄生虫益处 进化树 必备技能:提问 搜索 Endnote
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流快速解决科研困难,我们建立了“宏基因组”讨论群,己有国内外6000+ 科研人员加入。请添加主编微信meta-genomics带你入群,务必备注“姓名-单位-研究方向-职称/年级”。高级职称请注明身份,另有海内外微生物PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。
点击阅读原文,跳转最新文章目录阅读


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









