






点击蓝字 关注我们
猪场的日常职业暴露改变了人的皮肤微生物组和耐药组

iMeta主页:http://www.imeta.science
研究论文
● 原文链接DOI: https://doi.org/10.1002/imt2.158
● 2024年1月1日,华南农业大学孙坚、连新磊和波士顿儿童医院Alaric W. D’Souza等团队在iMeta在线联合发表了题为 “Daily occupational exposure in swine farm alters human skin microbiota and antibiotic resistome” 的研究文章。
● 本文设计了一项包含猪场工人和学生两种队列的研究,结果显示,携带抗生素耐药基因(ARGs)的微生物在猪场环境和工人皮肤之间的传播,并有可能以农场工人为媒介向社区中的人群传播。
● 第一作者:陈东瑞、程珂
● 通讯作者:孙坚(jiansun@scau.edu.cn)、连新磊(xinlei_lian@scau.edu.cn)、Alaric W. D’Souza(alaric.dsouza@childrens.harvard.edu)
● 合作作者:万磊、崔超月、李龚、赵东豪、于洋、廖晓萍、刘雅红
● 主要单位:华南农业大学动物疫病防控全国重点实验室;华南农业大学兽医学院/岭南现代农业科学与技术广东省实验室,国家兽医微生物耐药性风险评估实验室;华南农业大学广东省兽药研制与安全评价重点实验室;广西农垦永新畜牧集团有限公司
亮 点
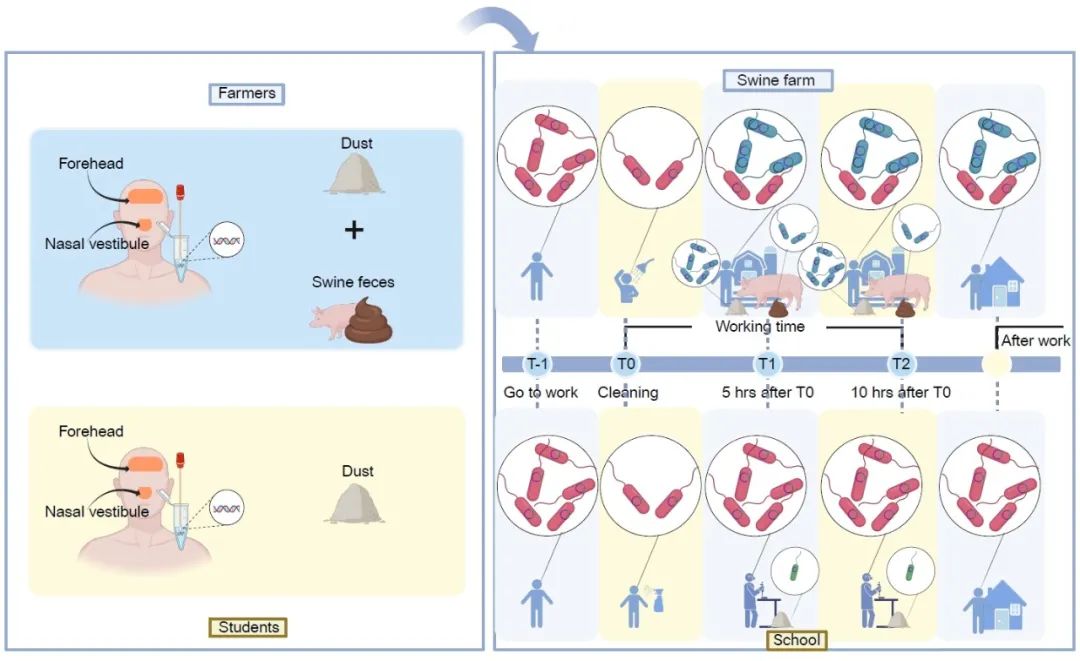
● 与学校环境相比,暴露于微生物更加丰富的养殖场环境将重塑人体皮肤微生物组和耐药组;
● 5小时的养殖场职业暴露足以改变工人皮肤微生物群和耐药基因结构;
● 微生物携带耐药基因在猪场环境和工人皮肤之间转移。
摘 要
抗微生物药物耐药性(AMR)是对全球公共卫生的主要威胁,携带抗生素耐药基因(ARGs)的微生物广泛分布于人类、动物和环境中。农业环境正在成为ARGs和抗生素耐药菌(ARB)的重点研究领域。虽然皮肤是ARGs和ARB的重要储存库,但两者在畜牧业环境和人类皮肤之间的传播机制尚不清楚。本课题组前期研究证实,长期的猪场环境暴露会改变人的皮肤微生物群,但变化的时间线尚未确定。为了确定影响发生的具体时间,本课题组设计了一项包含猪场工人和学生两种队列的研究,通过收集人的皮肤和所其处环境样本来探索猪场单个工作时长内的职业暴露对人体皮肤微生物组的影响。结果表明,与学校环境相比,短暂暴露于微生物更加丰富的养殖环境即可重塑人的皮肤微生物组和耐药组。猪场工人暴露于养殖环境5小时,其皮肤中的微生物组和ARG结构就足以被携带ARGs的养殖环境微生物所改变,并被保存了下来。数据表明,携带ARGs的微生物可能在猪场环境和工人皮肤之间传播,并有可能以农场工人为媒介向社区中的人群传播。以上结果引起了人们对ARGs可能向更广泛社区传播的担忧。因此,有必要在生产过程中采取相应的干预措施,减少养殖环境中ARGs和ARB传播的可能性。
视频解读
Bilibili:https://www.bilibili.com/video/BV1eT4y1p7eE/
Youtube:https://youtu.be/RyelJlCeTS8
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
全文解读
引 言
抗微生物药物耐药性(AMR)的出现和传播对全球公共卫生构成严重威胁,耐药基因(ARGs)广泛分布于人、动物和环境中。由于抗生素的滥用,家畜养殖环境正在成为ARGs和耐药菌(ARB)的重点研究领域。本研究关注畜牧养殖场环境暴露的科学依据主要基于以下原因,第一个原因是因为畜牧业代表了自然环境和人类之间的中间界面,阐明畜牧业如何在环境和人类微生物组之间架起桥梁具有重要意义。之前的报道强调,通过动物产品、环境与人类之间频繁的微生物交换是环境、动物和人类复杂相互作用的结果。第二个原因是畜牧业具有丰富的职业暴露微生物和非微生物因素。家禽、猪和牛养殖中丰富的微生物种类主要作为职业暴露的微生物因素,工人的皮肤微生物群可能很容易通过直接接触动物、铺垫材料、喂养设施,特别是空气中的微生物群落而受到环境微生物的干扰。已有研究报道养殖场,尤其是养猪场的空气中充满了大量的灰尘、细菌和真菌,空气中细菌的浓度可以达到2×107拷贝数/m3,比通常在室内空气中测量的水平高一倍。此外畜牧业一般要求较高的卫生标准,以最大限度地避免传染病造成的经济损失。大量使用消毒剂、抗菌剂和防腐剂,这可能会改变人类微生物组模式。第三个原因是由于畜牧业对抗菌药物的滥用给人类和自然微生物系统带来了巨大的压力,畜牧养殖农场环境中通常富集着大量的ARGs和ARB。研究ARGs和ARB在动物、人类和环境中的分布和传播模式,对减轻AMR的传播有重要意义。
皮肤是人体最大的器官,为微生物提供生态位,包括细菌、古生菌、真菌和病毒被统称为皮肤微生物组。皮肤上高丰度且长期定植的特定微生物处于长期平衡状态,丰度中等或低丰度微生物群则容易发生变化。皮肤微生物群落的组成和多样性受多种因素的影响,包括年龄、性别、遗传变异等内在因素,以及生活方式、日常行为和环境暴露等外部干扰因素。清洁和消毒等行为可以改变皮肤微生物群落组成,但主要影响暂驻型皮肤微生物。研究还表明,微生物群可以从环境转移到人体皮肤上。例如,与城市环境相比,生活在农村和森林环境附近的人皮肤上的微生物多样性更高。暴露于城市绿地可以通过改变人体微生物群组成和增加其皮肤和鼻腔中的微生物多样性来改善人类健康。Ahn等人在一篇综述中讨论了一些宏观环境因素,在这些类型中,与工作有关的职业暴露往往比较常见。职业性暴露会改变工人的皮肤微生物群,并可能威胁他们的健康。一项对罗马尼亚博物馆工作人员皮肤微生物组的研究显示,超过一半的研究对象皮肤真菌定植率较低,并且在他们的皮肤微生物组中检测到一些ARB。美国对农场工人皮肤微生物群的研究表明,频繁和动物进行肢体接触可能导致人类前臂皮肤微生物群的变化,增加农场工人皮肤感染异常病原体和皮肤疾病的风险。消毒卫生程序可能是生产过程中去除皮肤上暂驻型微生物的有效方法。由于非洲猪瘟病毒(ASFV)、肺炎支原体和猪链球菌等动物病原体在中国的流行,包括我们研究地点在内的大多数猪场要求工人在进入前采取消毒卫生措施来消除这些病原体的危害。
最近的一些研究关注了环境暴露对农场工人口腔或肠道微生物组和耐药组的影响,以及人体肠道和环境之间ARGs的传播。与更稳定的肠道或口腔微生物群落相比,人体皮肤微生物群具有更高的变异性,它也是重要的ARG储存库。畜牧业环境和人类皮肤之间ARGs和ARB传播的特征和程度在很大程度上是未知的。因此,猪场环境暴露对人体皮肤微生物组和耐药组的影响非常令人感兴趣。皮肤微生物组调查研究的一种常用方法是扩增子测序。该方法简化了计算分析,但也存在一些缺点。例如,可变区选择或引物选择可能对测序结果产生深远的影响。物种分类学研究的分辨率很低,大多数终止于属水平。详细的皮肤微生物学研究需要更准确和完整的微生物基因组信息,以便对不同菌株进行功能分析和物种分类。宏基因组测序可以更好地实现这些目标。
本研究中我们比较了学生和猪场工人的宏基因组测序样本,以了解猪场日常职业暴露对人皮肤微生物组和耐药组的影响。我们特别关注暴露时间,以阐明职业暴露后微生物群和耐药组在一天内变化的时间过程。我们的调查显示,暴露5小时足以改变工人皮肤中的微生物群和ARG结构。我们还探讨了ARGs在猪场环境和工人皮肤之间的传播机制。这些发现有助于深入了解ARGs向更广泛社区传播的潜在风险,并突出了公共和职业卫生干预措施的机会。
结 果
宏基因组测序和质量控制
本研究对猪场组和学校组共176份志愿者皮肤样品和128份环境样品进行宏基因组测序,以剖析短期猪场环境暴露前后皮肤微生物群和耐药基因组的动态变化。其中T0组(志愿者在完成清洁后再次取样)中所有样品的DNA产量都不能通过Qubit检测(在50μL洗脱的无菌水中<0.001 ng/μL),无法建库测序。另外有27个样本没有测序成功,共有121份人体皮肤和111份环境样本测序成功,可用于后续分析。这些测序样本的各项质量指标如表S3所示。
皮肤微生物群多样性在暴露后增加
为了了解日常职业暴露对健康个体皮肤微生物群多样性的影响,我们分析了来自工人和学生的前额皮肤(Fh)和鼻前庭皮肤(Ns)拭子样本的微生物群落。与暴露前相比,在猪场工作环境中暴露5小时后,Fh组和Ns组的微生物群落Alpha多样性(香农指数)极显著升高(p=0.0042)或呈现强烈的上升趋势(p=0.056),而暴露5至10小时后,微生物群落多样性没有发生显著变化(Fh: p=0.92,Ns: p=0.53)。在学校组中,学生皮肤微生物群多样性在一天内保持稳定(图1B和图S1)。
采用非度量多维尺度(NMDS)和置换多变量方差(PERMANOVA)分析对工人和学生的皮肤微生物群落结构进行了表征(图1C)。在工人中,从时间点T-1 (T-1)到时间点T1 (T1),Fh组微生物群落结构变化显著(R2 = 11.8%, p = 0.01),从时间点T1到时间点T2 (T2),Fh组微生物群落结构变化不显著(R2 = 6.9%, p = 0.169)。Ns组微生物群落结构从T-1到T1变化不大(R2 = 6.2%, p = 0.260),从T1到T2几乎无变化 (R2 = 1.5%, p = 0.943)。在学生中,从T-1到T1, Fh组和Ns组的微生物群落结构变化不大(Fh: R2 = 5.5%, p = 0.321;Ns: R2 = 4.1%, p = 0.536),从T1到T2均几乎无变化(Fh: R2 = 3.6%, p = 0.610;Ns: R2 = 1.7%, p = 0.928)。这些结果表明,暴露在猪场环境中的工人皮肤微生物群落可以迅速发生变化并达到稳定状态。相比之下,学生的皮肤微生物群落结构在一天内没有明显变化。
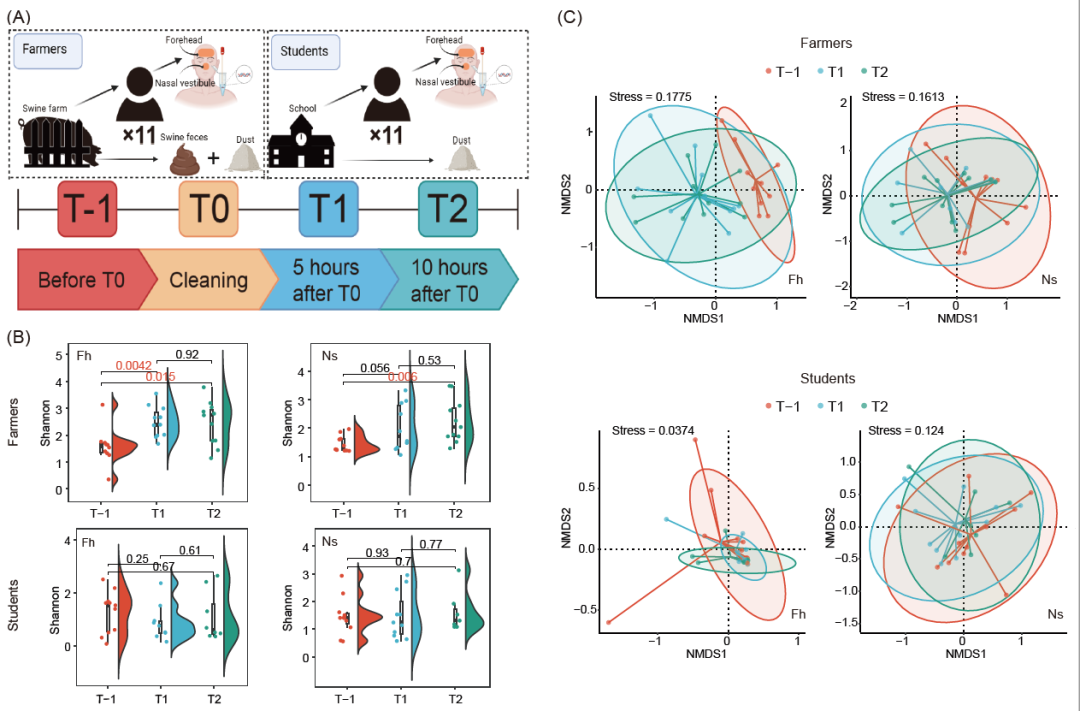
图1. 日常职业暴露后皮肤微生物群多样性的变化
(A) 实验设计示意图:农场工人,11名养猪场工人;学生,11名大学生;T-1:暴露前,T0:皮肤清洁,T1:暴露5h后, T2:暴露10h后。(B) 两个队列(农场工人和学生)中Fh组和Ns组样品在T-1、T1和T2时的微生物多样性(香农多样性指数)的变化。用红色标记的p值小于0.05。(C) 基于T-1(红色)、T1(蓝色)和T2(绿色)的Bray-Curtis不相似性,对两个队列中Fh组和Ns组的微生物群落结构进行非度量多维尺度(NMDS)分析
猪场皮肤微生物群组成变化与职业暴露相关
为了解图1显示的α多样性和组成结构变化背后的具体物种分类变化,我们在门、属和种三个分类介元上研究了工人皮肤微生物群。我们的物种分类分析结果鉴定出25个门,391个属,1189个种的古细菌,细菌和真菌(表S4)。
职业暴露前,工人前额皮肤微生物群以放线菌门为主,鼻前庭皮肤微生物群以厚壁菌门为主,分别占61.09%和49.35%。如图2D和表S5所示,优势门(丰度总和前三名)暴露前后几乎不变,只有来自鼻前庭皮肤的变形菌门有显著的相对丰度变化(T1/T-1: p < 0.05)。
三元图(图2A)显示了不同时间点工人皮肤微生物群在属水平上的相对丰度。总体而言,优势属主要来自放线菌门(Actinobacteria)、厚壁菌门(Firmicutes)和变形菌门(Proteobacteria)三个门,暴露后优势属变化不大,在T-1时优势属主要来自放线菌门(Actinobacteria),而在T1和T2时优势属主要来自放线菌门(Actinobacteria)和变形菌门(Proteobacteria)。对于每种样本分组,使用Spearman相关系数来描述属之间的邻域关系。通过比较不同时间点上Fh组和Ns组皮肤微生物群的相关性共发生网络(图2B)发现,与T-1相比,两组在T1时的属间相关性较弱,网络密度(用于描述微生物之间潜在连接的部分)和网络集中度(用于衡量网络中所有节点中心性得分与网络中获得的最大中心性得分的分散程度)急剧下降。我们使用线性判别分析(LEfSe)来确定职业暴露前后特征微生物属的变化。发现Fh组和Ns组在T1时乳杆菌、短杆菌和肠球菌等10个属的相对丰度均显著高于T-1时(图2C)。
前额皮肤发生显著变化的物种为阿尔特氏葡萄球菌、柯氏葡萄球菌、松鼠葡萄球菌和干燥棒状杆菌,鼻前庭皮肤发生显著变化的物种为阿尔特氏葡萄球菌、松鼠葡萄球菌和干燥棒状杆菌。在T-1时的低丰度物种(小于0.5%)的相对丰度发生了显著变化,而高丰度物种保持稳定。种水平的物种分类注释结果如表S7所示,并在堆叠面积图中显示(图2D)。
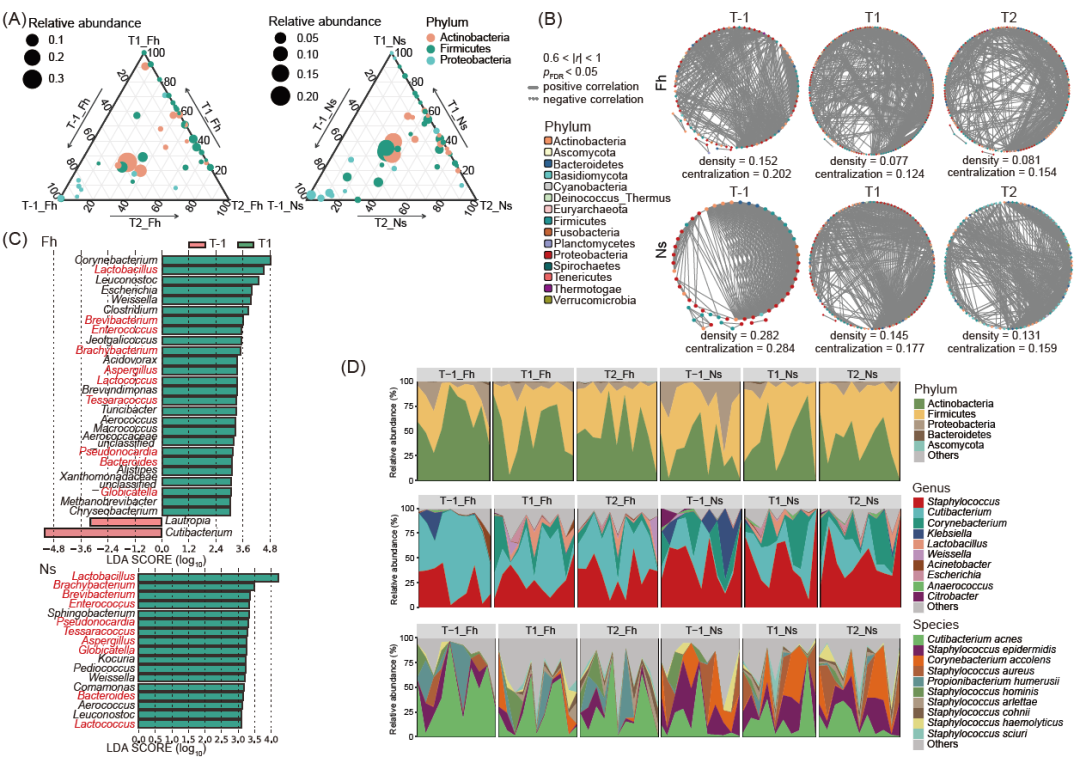
图2. 工人皮肤微生物群组成在不同时间点的变化
(A) 三元图显示了Fh和Ns组样品在T-1、T1和T2期间的微生物群组成丰度变化。点的大小表示样本中属的平均丰度,点的颜色表示其对应的门。(B) 暴露后,属水平上的微生物共生网络显示出属间相关性降低。(C) T-1和T1时特征微生物属的LEfSe分析。仅显示线性判别分析(LDA)评分(log10) > 3的结果。(D) 按门、属和种水平展示微生物相对丰度的堆叠面积图。x轴上被省略的标签是样品名称。
猪场环境和工人皮肤之间的微生物传播
通过暴露前/后皮肤微生物种类的差异性分析,从前额皮肤样品中鉴定出41种发生显著变化的菌种,从鼻前庭皮肤样品中鉴定出12种显著变化的菌种,包括干燥棒状杆菌、无枝菌酸棒杆菌和缓慢葡萄球菌等(图3A和表S8)。其中有几种是条件致病菌,除血链球菌外,其余均在暴露5小时后富集。这些结果与我们的发现一致,即环境暴露导致工人皮肤微生物群落的丰富度和均匀性发生变化。暴露前和暴露后显著不同的物种中有11种为Fh组和Ns组共有。在日常职业暴露后,这些物种的相对丰度在前额皮肤微生物组中的变化大于鼻前庭皮肤组(图S2A)。虽然这些变化的物种在个体水平上的相对丰度占比不同,但在T-1时均小于0.5%,并且由于职业暴露导致的变化趋势大致一致(图S2B)。
此外,图3B显示了短期暴露后环境和工人皮肤共有的微生物种类数量的变化情况。从T-1到T1,工人皮肤与环境微生物群共享的物种显著增加,工人与灰尘微生物群共享的物种数量始终远远大于其与粪便微生物群共享的物种数量。Fh组与灰尘共有物种数从58种(7.7%)增加到289种(38.6%),Ns组与灰尘共有物种数从41种(6.9%)增加到214种(35.5%)。微生物溯源分析评估皮肤微生物与环境微生物之间的关联(图3C)显示,T-1时,29.88%的前额皮肤微生物组成与灰尘微生物相关。这一比例在T1和T2分别增加到42.00%和48.69%。对于鼻前庭皮肤样本,在T-1时,19.21%的微生物组成与灰尘微生物相关,在T1和T2时这一比例分别增加到48.95%和49.90%。各时间点粪便微生物对皮肤微生物组成的影响最小,除T1时的鼻前庭皮肤外,各组的比例均小于1%。皮肤微生物中仍有部分源与本次研究所涉及的因子无关,其是否来源于猪场的其它环境因子仍有待探究。通过研究灰尘样品的微生物组成,发现其中相对丰度排名前15的物种中,有9种是上述结果中的差异物种(图3D)。这些结果表明,在环境对皮肤微生物群的影响中,灰尘微生物比猪粪发挥更重要的作用。
为了更深入地了解工人皮肤变化的微生物群与环境微生物群之间的潜在共性,我们研究了菌株水平的种群结构。发现在上文火山图的结果中有8个物种共存于皮肤和环境样品(主要是环境暴露后的皮肤和灰尘样品)中,且存在密切的系统发育关系(标准化系统发育距离不超过0.1)(图S3和S4)。其中干燥棒状杆菌最为常见(图3E),它是一种存在于人和动物皮肤粘膜中的机会性病原体,能够引起心内膜炎、皮肤感染和其他疾病。
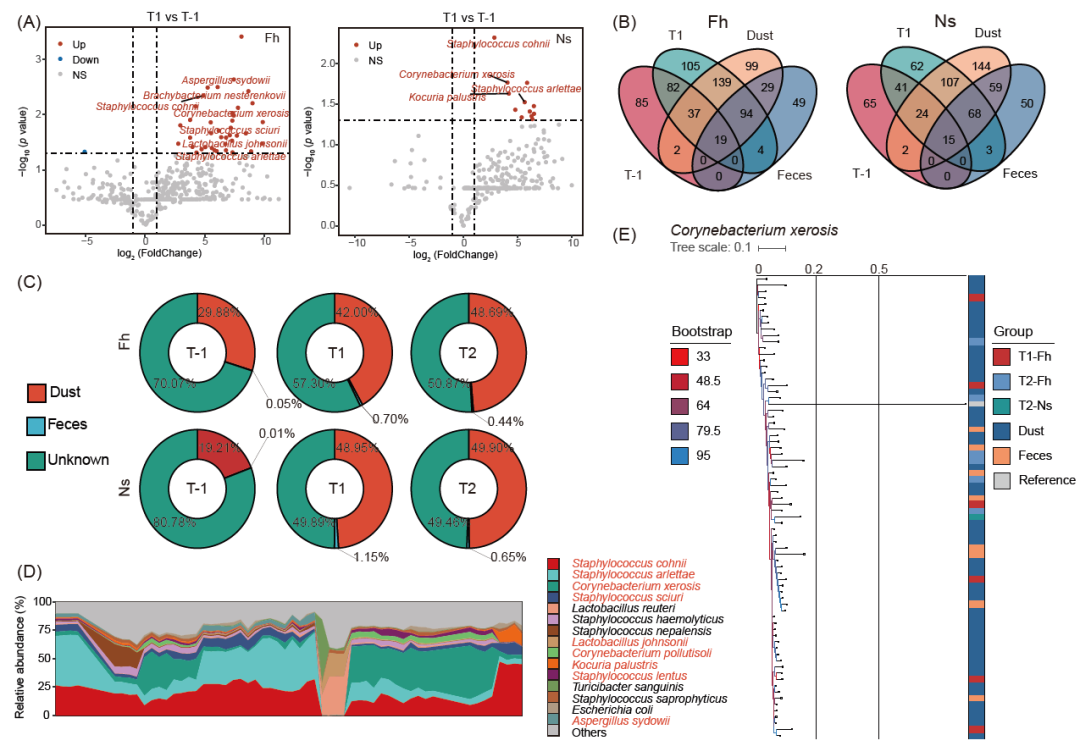
图3. 猪场环境和工人皮肤之间的微生物传播
(A)火山图显示职业暴露5小时后额头和鼻前庭皮肤在种水平上微生物群组成的变化,x轴表示log2(Fold Change), y轴表示- log10(p值)。红色为T1时显著增加的物种,蓝色为显著减少的物种。横线和虚线表示筛选标准(绝对差异倍数变化(FC)≥1.0, p值< 0.05)。(B)量化灰尘样品与前额和鼻前庭皮肤样品之间共有微生物种类的维恩图。(C) T-1、T1和T2三个不同时间点皮肤微生物群来源预测。(D)堆积面积图,显示前15种相对丰度最高的灰尘微生物。在相对丰度排名前15位的物种中,有9种是我们之前发现的皮肤上变化显著的物种。x轴上被省略的标签是样品名称。(E)利用StrainPhlAn3软件在菌株水平上构建的干燥棒状杆菌系统发育树。干燥棒状杆菌的参考基因组来自干燥棒状杆菌ASM364124v1。引导支持值由图例的颜色表示。
环境变化对皮肤耐药组结构的影响
为了评估ARGs的变化是否伴随着微生物群的变化,对工人皮肤耐药基因组进行了宏基因组分析,检测到20种ARG类型,它们包括了618种ARG亚型(表S9)。
与T-1相比,皮肤耐药基因组在T1时具有显著更高的香农多样性(Fh: p = 0.016, Ns: p = 0.023),但从T1到T2保持稳定(Fh: p = 0.99, Ns: p = 0.96)(图S5)。NMDS和PERMANOVA分析显示,从T-1到T1,皮肤耐药组的结构发生了显著变化(Fh: R2 = 24.2%, p = 0.01;Ns: R2 = 8.9%, p = 0.033), T1到T2则无显著变化(Fh: R2 = 3.9%, p = 0.624;Ns: R2 = 2.0%, p = 0.945)。暴露后的皮肤与灰尘耐药基因组重叠在一起,与猪粪耐药基因组部分重叠,提示皮肤与灰尘耐药基因组结构分布比较相近,与猪粪存在一定差异。学生皮肤中耐药组的多样性在一天内没有显著变化。总体而言,皮肤ARG多样性与其微生物组多样性发生的变化比较相似。
与暴露前相比,相对ARG丰度的总和(参考序列每千碱基的平均读数,RPKM)在额头皮肤中急剧增加(p = 0.0048),平均增加127.14%(图4A),在鼻前庭皮肤中略有增加(p = 0.15),平均增加4.01%(图S6A)。与暴露前相比,暴露后前额皮肤中ARG类型丰度增加最显著的是林可酰胺类,其次是恶唑烷酮类和磺胺类,ARG类型没有发生丰度显著降低的(图4B)。鼻前庭皮肤ARG类型无明显变化。为了确定丰度发生显著变化的ARG亚型,我们进行了暴露前/后的差异分析。暴露5小时后显著富集的ARG亚型主要为四环素类耐药基因,包括tet(X)、tet(X3-X6)、tet(W)等25个亚型,其次是氨基糖苷类耐药基因,包括AAC(6’)_Ie_APH(2’_Ia)、ANT(9)-Ia、aadA2等21个亚型和Macrolide - Lincosamde - StreptograminB (MLSB)类耐药基因,包括ErmB、ErmF、ErmT等13个亚型(图4C、S6B和表S10)。

图4. 日常职业暴露对皮肤耐药组结构的影响
(A) T-1、T1和T2时间点前额皮肤抗微生物药物耐药基因(ARG)丰度总和。箱体表示工人样本的分布。标红的p值小于0.05。(B) 前额皮肤ARG类型相对丰度的比较。统计采用学生t检验和Benjamini-Hochberg FDR校正。(C) 火山图显示职业暴露5小时后前额皮肤ARG亚型分布的变化。用log2FoldChange表示T1时ARGs的变化。红/蓝点表示ARGs明显增加/减少。用文字标记的圆点表示Fh组和Ns组在T1富集的共有ARGs。(D) 桑基图显示了不同时间点前额皮肤样本的ARG组成与环境样本的ARG组成之间的相关性。矩形的高度和颜色的深度都表明了人类皮肤微生物组与猪场环境微生物组之间的Spearman相关性。(E) 前额皮肤和环境样本中微生物组和耐药组的Procrustes分析。
猪场环境与工人皮肤之间ARGs的转移
工人皮肤与猪场环境样品耐药基因组的相关性在暴露5小时后显著增强,5小时至10小时保持稳定(图4D和图S6C)。其中,T-1时额头皮肤与灰尘的相关系数为r = 0.35,与粪便的相关系数为r = 0.23; T1时,额头皮肤与灰尘的相关系数为r = 0.80,与粪便的相关系数为r = 0.67; T2时,前额皮肤与粉尘的相关系数为r = 0.80,与粪便的相关系数为r = 0.63。Ns组的结果与Fh组基本相同,这表明灰尘中的ARGs可能在皮肤耐药组的变化中起重要作用,与微生物组的结果相似。基于Bray-Curtis距离的普鲁克(Procrustes)分析揭示整个耐药基因图谱和微生物群落组成有良好的拟合关系且显著相关(Fh: M2 = 0.2010, p = 0.001;Ns: M2 = 0.1388, p = 0.001)(图4E和图S6D)。这些结果表明,工人皮肤耐药组与其微生物组的变化趋势是一致的,这意味着ARGs的变化可能来自于微生物的变化。进一步分析支持这一假设, 我们对ARGs与暴露后发生显著变化的微生物进行Spearman相关分析,选择极显著和强相关(0.8 < |r| < 1, p < 0.05)关系,结合Network方法展示共发生网络。如图5A所示,皮肤和环境之间共存8种暴露后显著富集的微生物,这些微生物也是显著富集ARGs的潜在宿主。为进一步验证工人皮肤与环境中是否存在共享携带ARGs的宿主,本研究通过宏基因组分箱方法重组了64个环境样品的基因组,共创建出2338个MAGs。经过对其完整度和污染度的筛选,总共留下581个完整性≥50%和污染度≤10%的MAGs。对这些MAGs通过GTDB-TK软件进行物种注释,并进行系统发育树分析,发现它们属于10个细菌门,其中最丰富的是厚壁杆菌门(58.7%),其次是放线杆菌门(20.7%),拟杆菌门(13.1%)和变形杆菌门(3.6%)(图5B)。581个MAGs中有455个被成功注释到种水平,包括干燥棒状杆菌(n=29)、阿尔莱特葡萄球菌(n=21)、松鼠葡萄球菌(n=14)、约氏乳杆菌(n=8)和涅氏短状杆菌(n=3),它们都是本研究中暴露后在皮肤上发生显著富集且共存于环境样品中的差异菌株。对这些MAGs进行耐药基因注释,共筛选出76个携带多种ARGs的宿主,它们携带的ARGs中有15种是显著富集ARGs,如sul1、optrA、poxtA和tet(X)等。分箱分析部分结果与上文网络分析得到的结果一致:阿尔莱特葡萄球菌是tet(44)、optrA、spd、ARL-1、ErmT、ANT(9)-Ia和ANT(6)-Ib基因的潜在宿主,干燥棒状杆菌是cmx和APH(4)-Ia基因的潜在宿主。松鼠葡萄球菌是ANT(9)-Ia基因的潜在宿主。这证明了本研究耐药基因宿主定位的可信度与准确度。
如之前的研究所做,将同时具有ARGs和MGEs的contigs归类为移动ARG。然后在ARGs和MGEs之间采用5 kb的长度限制,以将分析限制在位于MGEs附近的ARG上,在环境暴露后工人皮肤富集的ARGs中发现25种在皮肤和环境样品中共存的具有相同模式的移动ARGs,如AAC(6’)-Ie-APH(2’’)-Ia、poxtA、sul1和tet(X)等(图5D和表S2)。AAC(6’)-Ie-APH(2’)-Ia基因编码aac(6’)-Ie-aph(2’)-Ia酶,该酶对庆大霉素和除链霉素外的几乎所有临床氨基糖苷类药物都具有耐药性。移动AAC(6')-Ie-APH(2 ')-Ia具有两种不同的移动ARG模式:(i)插入序列IS256;(ii)插入序列IS431R。poxtA基因是一种可转移的恶唑烷酮耐药基因,据报道,该基因与用于治疗畜禽呼吸道和肠道细菌感染的氟苯尼考残留有关。移动poxtA基因有三种不同的移动ARG模式:(i)插入序列IS1216E;(ii)插入序列ISS1W和(iii) ISLgar4。sul1和tet(X)是猪场常见的兽药ARG,其中移动sul1基因与4种模式相关,是最多样化的移动ARG。
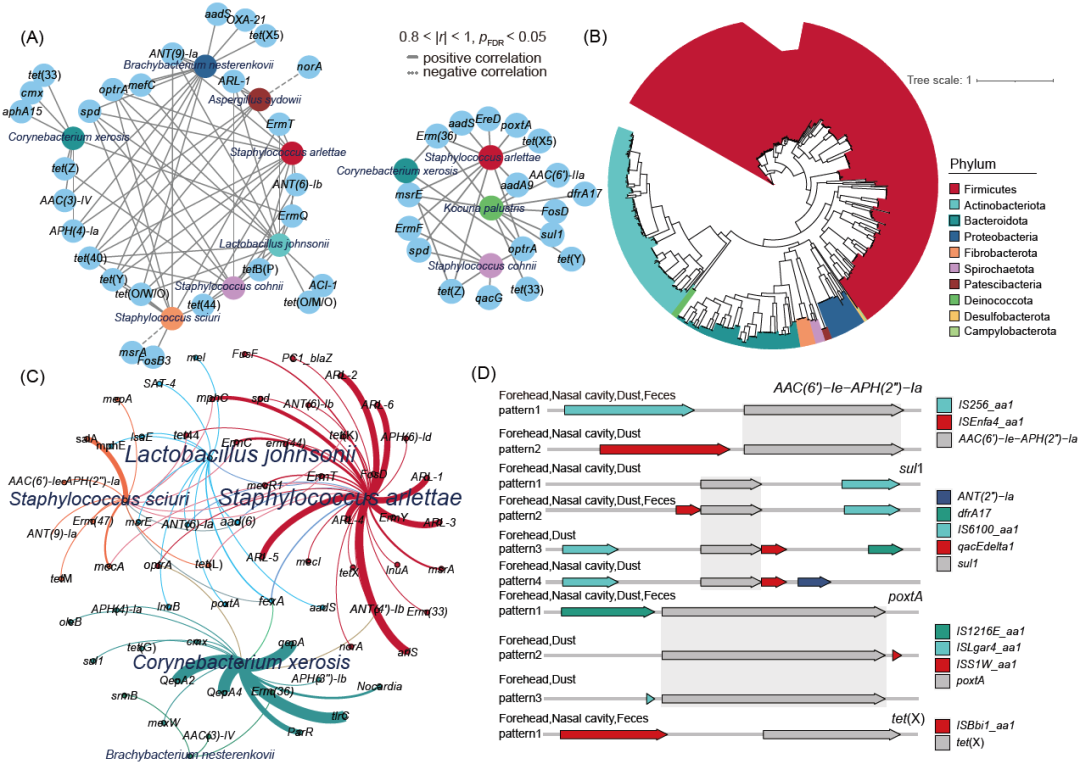
图5. ARGs通过微生物在猪场环境和人皮肤之间传播
(A) 发生显著变化的ARGs与环境传播的微生物共发生网络分析(p < 0.05,绝对相关系数> 0.80)。(B) 宏基因组组装基因组(MAGs)的系统发育分配树。树是根据门来着色的。(C) 主要MAGs和显著差异ARGs之间的共现模式。节点根据物种着色。节点大小与连接数成比例(平均加权)。曲线的宽度代表了宿主携带ARG的丰度。(D) 移动ARGs基因组线性排列分析。四个ARGs AAC(6’)_Ie_APH(2’)_Ia, sul1, poxtA,和tet(X)在图中被展示,ARGs用灰色阴影表示。
方 法
研究设计和样本收集
我们进行了一项纵向队列研究,以调查日常职业暴露对农场工人皮肤微生物组和耐药组的影响。本研究以某养猪场为试验组,以某学校为对照组。养猪场组(队列ID: Workers)的志愿者(年龄= 45±10岁,字母:A、B、C、D、E、F、G、H、I、J、K)为具有1-2年工作经验的广西某养猪场一线工作人员,该养猪场采用封闭式管理模式,平均自繁种猪约2万头。学校组(队列ID: Students)的志愿者(年龄= 25±3岁,字母:L、M、N、O、P、Q、R、S、T、U、V)为华南农业大学学生。农场的生物安全措施要求工人在进入农场前进行彻底消毒,为了设计一个基于实际生产过程的研究,对照组的学生也被要求与农场工人同时彻底清洁皮肤和鼻腔并取样。因此,在四个时间点T-1(上午8点:工人进入农场前),T0(皮肤清洁),T1 (T0后5小时)和T2 (T0后10小时),同时从两个队列志愿者中收集前额皮肤拭子和鼻前庭皮肤拭子。猪场工人提供了生产操作区和通风口的灰尘样本,以及分娩和妊娠室的猪粪便样本。学校的灰尘样本是从学生经常工作的实验室和自习室收集的。每个采样区收集大约3到5个样本。
在收集皮肤样本之前,我们获得了志愿者的知情同意,向他们声明我们的试剂是安全的。我们还收集了志愿者的个人情况和健康史信息,包括年龄、工作时抗微生物药物暴露、皮肤健康状况和其他相关因素。表S1详细列出了这些信息。
使用的皮肤样本拭子采集技术在我们之前的研究中有描述。灰尘样品的采集方法是用消毒过的生理盐水浸湿无菌棉签,然后在采样表面反复擦拭,收集样本。猪粪便标本的收集方法是用无菌勺子切掉粪便的中间部分。在提取DNA之前,所有样品在-20°C下保存。
DNA提取
根据制造商的说明,使用QIAamp PowerFecal Pro DNA试剂盒(Qiagen, Germany)从所有样品中提取基因组DNA。首先使用NanoDrop 2000分光光度计(Thermo Fisher Scientific, USA)粗略测定环境样品基因组DNA的纯度和浓度。在1%琼脂糖凝胶上监测DNA降解和污染情况,使用Qubit®DNA Assay Kit,在Qubit®3.0 Flurometer (LifeTechnologies, USA)上测量DNA精确浓度。皮肤拭子样本的DNA浓度一般较低,只需要测量精确的浓度。
宏基因组测序和质量控制
测序前构建了一个包含350bp DNA片段的文库。将每个样品在Illumina HiSeq 4000平台上使用PE150(双端测序)策略进行测序。将每个样品的原始测序读数使用Fastp (v0.19.7)独立处理,以进行质量控制。
去除宿主DNA
使用Bowtie2 (v2.4.5)建立猪和人参考基因组数据库,使用猪参考基因组Sscrofa11.1 (NCBI登录号GCA_000003025.6)和智人参考基因组GRCh38。p13 (NCBI登录号GCA_000001405.28)。使用KneadData (v0.10.0)将质控后的clean reads与参考基因组数据库进行比对,去除宿主DNA污染。
宏基因组数据物种分类注释和耐药基因的量化
使用MetaPhlAn (v3.0.14) 对去除宿主DNA的clean reads在细菌、古细菌和真核生物界进行物种组成注释,准确估计出每个样品在界、门、纲、目、科、属和种的分类学上的物种相对丰度。使用ShortBRED (v0.9.3)通过综合抗生素耐药性数据库(CARD, v3.2.4, 2022年7月下载)对ARGs的相对丰度进行量化。如果ShortBRED有效比对结果低于2或每百万样品读数中参考序列的平均读数(mean reads per kilobase of reference sequence per million sample reads,RPKM)低于0.001,则被过滤掉。
共有菌株和移动ARGs分析
采用StrainPhlAn3.0进行菌株水平分析和菌株跟踪分析。将得到的比对结果作为IQtree (v2.2.0.3)的输入,设置模型选择和10000次超快自举重复构建系统发育树。所有得到的系统发育树在iToL中绘制。所有参考基因组均来自NCBI网站(https://www.ncbi.nlm.nih.gov/)。使用MEGAHIT (v1.2.9)对序列进行组装。在GitHub (https://github.com/dantaslab/resAnnotator)上可以找到ARG注释器(resAnnotator.py)管道,用于对获得的contigs进行耐药基因注释,我们使用的耐药基因注释数据库是CARD数据库。提取并过滤携带ARGs的contigs(大于500bp)。使用BLASTP将这些contigs的ORF序列与ISfinder数据库进行匹配。ARG和MGE之间距离大于5 kb与ARG和MGE序列重叠的contigs都被丢弃。剩下的contigs被认为是移动ARGs。表S2总结了移动ARG各种模式的结构(MGE类型、ARG类型和长度等)。对于志愿者皮肤和环境中共存的移动ARG,使用R软件包“gggenes”对基因结构进行可视化。
宏基因组分箱和MAGs的耐药基因注释
使用MetaBAT2 (v2.12.1)重构猪场环境样本基因组。使用dRep (v3.4.0)以95%的平均核苷酸同一性(ANI)去冗余,每个MAG在分类学上相当于一个微生物物种。使用CheckM (v1.2.1)对所有MAGs的完整性和污染度进行评估,并使用完整性≥50%和污染度≤10%进行过滤。使用Genome Taxonomy Database Toolkit (GTDB-Tk) (v2.1.0)对保留的MAGs进行注释,并使用BLASTX在CARD数据库中进行比对。
统计分析
采用R平台(v4.1.2)进行统计分析。为了探究暴露后皮肤微生物组是否发生显著变化,我们使用“vegan”包根据样品不同分组的Bray-Curtis距离进行非度量多维排列(Non-metric multidimensional scaling,NMDS)分析,观察其结构差异性。置换多元因素方差分析(Permutational multivariate analysis of variance,PERMANOVA)被用来确定环境暴露对皮肤微生物群以及抗生素抗性组的重要性。利用Galaxy网站(http://huttenhower.sph.harvard.edu/galaxy/, v1.0)对特征微生物类群进行LEfSe分析。快速准确的微生物来源追溯工具(Fast expectation–maximization microbial source tracking, FEAST)被用于识别细菌群落的来源,用R包“FEAST”实现。我们使用“vegan”包对皮肤微生物组和抗生素抗性组进行普鲁克(Procrustes)分析,首先对这两个数据集进行Bray-Curtis距离转换,之后进行PCoA降维排序,用“protest”函数估计Procrustes统计量(从对称的Procrustes偏差平方和中得到的类似于相关性的统计量)的意义,并对偏差平方和M2进行置换999次的普氏检验。共发生网络图可视化是在Cytoscape(版本号;3.7.0)的互动平台上进行的。分别在Gephi (v0.9.7)交互平台和iToL在线网站上绘制MAGs的ARG注释网络可视化和系统发育树图。其他图均使用R包“ggplot2”绘制。经Benjamini-Hochberg校正后,p值(使用Student's t检验)< 0.05具有统计学意义。
讨 论
健康人体皮肤微生物群落的主要门类为放线杆菌门、厚壁菌门、变形杆菌门和拟杆菌门。检测到本研究中志愿者皮肤微生物群落暴露前/后都是由这四个门类主导,和之前的研究结果相似,验证了所采用方法、所收集标本的代表性和合适性。总体来说,暴露后额头与鼻前庭皮肤微生物群的变化趋势基本一致,鼻前庭皮肤的变化略微保守,可能是因为其只部分暴露于外界环境中,比额头皮肤更稳定。农场工人皮肤微生物群的结构和多样性在职业暴露5小时后发生了一定变化,且达到了稳定状态,而学生皮肤微生物群则全天保持稳定。这说明猪场的日常职业暴露动态地改变了人类皮肤微生物组。与其他环境相比,畜牧业环境富含多种微生物,即使只暴露在其一天也会引起人体皮肤微生物组的改变。LEfSe分析结果显示,暴露5小时后LDA评分最高的3个特征属分别是乳酸杆菌属、短杆菌属和肠球菌属。肠球菌属和乳酸杆菌属在人和动物肠道菌群中普遍存在,因此在被人畜粪便污染的环境中也很常见。短杆菌属经常在各种环境的灰尘中发现,因此通过日常工作或灰尘传播,在工人皮肤中发现这些属的富集并不奇怪。暴露后,干燥棒状杆菌等条件致病菌也显著富集。干燥棒状杆菌存在于人和动物的皮肤粘膜中,可引起心内膜炎、皮肤感染等疾病。无枝菌酸棒杆菌是一种潜在的多重耐药机会性病原体,也是会导致人类和动物严重感染的病原体,特别是当涉及免疫功能低下患者的鼻腔环境时。缓慢葡萄球菌是一种可引起鼻窦炎的鼻腔病原体。然而,我们也检测到一些有益菌,如科氏葡萄球菌是一种潜在的生物治疗性皮肤共生菌,可以减轻皮肤炎症。微生物溯源结果发现,暴露5小时后额头和鼻前庭皮肤微生物来源于灰尘占比和与灰尘微生物群落共享的物种数目均显著升高,而来源于猪粪的占比始终微乎其微,可以说明灰尘对导致工人皮肤微生物发生变化的贡献远高于粪便。一项研究有类似结果,室内灰尘微生物群落可以与人体皮肤微生物群落相互转移。动物粪便是空气气溶胶中ARGs和病原菌的主要来源,但是由于大气扩散作用,携带ARGs和微生物的气溶胶可以传播至周围十公里,也可以积累沉降在灰尘中,与工人接触后转移到其皮肤上。这可能是来源于直接采集的猪粪微生物占比较低的原因,后续的研究可能需要采集空气样品进一步确定猪粪微生物对皮肤微生物变化的贡献。微生物溯源和菌株系统发育关系分析结果证明了工人皮肤和他们周围的灰尘之间可能存在着广泛的微生物交换。综上所述,可以推测工人进入猪场工作一段时间后,皮肤和灰尘环境之间开始进行微生物交换,从环境暴露5小时开始,猪场环境中的微生物可以稳定停留在工人皮肤,至其工作结束返回居住地(T2)。
在之前关于猪场环境暴露对人肠道微生物组的影响研究中,ARGs与环境介导的微生物传播事件全面伴随转移。本研究中结果亦是如此,职业暴露后皮肤ARGs多样性发生的变化与其微生物群的变化趋势保持一致,工人皮肤耐药基因组与其微生物组显著相关,先前的研究报道,在猪场职业暴露后,人类口腔和肠道中ARGs的相对丰度有所增加,我们的结果也类似,特别是在前额皮肤中,ARGs的相对丰度在暴露5小时后增加了一倍,此后保持稳定。这表明猪场日常职业暴露可引起皮肤中ARGs的富集,这也表明工人和猪场环境之间可能存在ARGs的水平转移,暴露后皮肤中ARGs的组成趋向于灰尘耐药组。结合上文中的微生物溯源结果,可以推测变化的ARGs与工人皮肤和环境之间广泛交换的微生物有关。环境暴露5小时后工人皮肤富集的ARG亚型主要属于四环素类、氨基糖苷类和MLSB类耐药基因,这几种ARG类型被报道常见于畜牧养殖场动物粪便和灰尘中,例如有研究分析中国大型猪场新鲜猪粪样品中的抗生素抗性组,结果发现猪肠道中氨基糖苷类、MLSB类和四环素类的ARGs丰度最高。值得注意的是,暴露后显著富集的ARGs中出现了cfr(A),optrA和poxtA这三种基因,均为苯尼考尔-恶唑烷酮-四环素(phenicol-oxazolidinone-tetracycline)抗性基因,它们的出现成为公共卫生的重大挑战,因为不仅降低了对最后手段药物利奈唑胺的敏感性,而且还降低了对兽药中广泛使用的氟苯尼考和多西环素的敏感性。据报道这些基因在猪场动物粪便的肠球菌分离株中普遍存在。而本研究采样的猪场有泰乐菌素(大环内酯类抗生素)、庆大霉素(大环内酯类抗生素)、阿米卡星(氨基糖苷类抗生素)、氟苯尼考(广谱抗生素)和四环素等以上提到抗菌药物的使用史。因此可以推测在我们采样的猪场,大量的抗菌药物被用于饲料添加剂,导致ARGs和ARB在猪肠道中富集并随其粪便排出,在移动动物和处理粪便过程中被气溶胶化逸散到空气中或沉降并积累在灰尘中,在接触猪场环境后,工人皮肤上出现了这些ARGs的富集。有对广西省某猪场环境中的空气传播细菌群落和耐药基因组的研究指出,猪场空气中的耐药基因组主要对四环素类、氨基糖苷类和林可酰胺类产生耐药性。与本研究结果相似,也进一步证明了这一观点。值得注意的是,我们在职业暴露后富集的ARGs中鉴定出tet(X)基因及其变体tet(X3)、tet(X4)、tet(X5)和tet(X6)。替加环素是治疗多重耐药细菌感染的最后手段,tet(X3)、tet(X4)和tet(X5)可显著降低替加环素的治疗效果。虽然替加环素不用于畜牧业,但滥用四环素作为动物饲料添加剂促进了tet(X)变体的出现。tet(X)可通过细菌克隆传播,不同的变体通常具有不同的潜在细菌宿主。研究表明tet(X)变体在人类和牲畜环境中都有出现。tet(X)变体的广泛传播将导致抗菌药物失效,会对未来人类健康存在严重威胁,因此监测人类、动物和环境中的tet(X)及其变体具有重要意义。tet(X)基因和其变体的富集可能与采样猪场四环素类药物的使用频率较高有关,提示畜牧养殖业需要进一步规范合理养殖。
抗菌药使用对人类健康的影响取决于农场和人类相关微生物群之间的连通性。这种连通性既包括动物和人类相关细菌之间横向ARGs转移的潜力,也包括动物和农场环境中的ARB向人类宿主的传播性。我们的分析表明,职业暴露后的皮肤耐药组和灰尘耐药组之间存在高度的相关性,这说明ARGs可能是从导致微生物群变化的环境中获得的。Spearman相关性分析绘制共发生网络图结果发现暴露后丰度显著升高且在工人皮肤与环境中共存的微生物全部为携带显著富集ARGs的潜在宿主,但需要进一步验证。ARGs的捕获、富集以及传播在很大程度上依赖可移动遗传元件(Mobile genetic elements,MGEs)的作用,有研究表明MGEs向多种细菌(包括病原体和人类共生菌)的转移已被确定为ARGs可以持久性传播的主要原因。基于此,本研究对携带ARGs的contigs进行MGEs注释,发现有17.40%中同时存在MGEs序列,且在环境暴露后工人皮肤丰度显著升高的ARGs中发现25种在皮肤和环境样品中共存的具有相同模式的移动ARGs,如AAC(6')-Ie-APH(2'')-Ia、sul1、poxtA和tet(X)基因等,通过分析发现它们的遗传环境相似,这为工人皮肤和猪场环境微生物群之间共享ARGs提供了证据。其中观察到poxtA基因在工人皮肤和环境中以移动ARG的形式出现,并在其周围发现了插入序列IS1216E,ISS1W和ISLgar4,已有研究证明IS1216E介导的转位和易位过程有助于poxtA基因在宿主微生物中的传播和持续存在,而IS1216E-poxtA模式的移动ARGs是最丰富的,在皮肤和环境总共21份样品中出现。
本研究尝试通过对含有ARGs序列的contigs进行物种注释的方法来获得携带ARGs的宿主微生物,然而由于超过半数的contigs未能准确注释到种,需要通过宏基因组分箱的方法获得MAGs,再对其进行耐药基因与物种注释,以此来获得两者之间直接的相关性。环境样品注释到种水平的MAGs中出现了5种暴露后丰度显著升高且共存于环境样品中的差异菌株,证实这些显著变化的微生物确实来源于环境。对这些MAGs进行耐药基因注释,共筛选出76个携带多种ARGs的宿主微生物,它们携带的ARGs中有15种是暴露后发生富集的ARGs,同时分箱结果与共发生网络结果部分一致,如阿尔莱特葡萄球菌和干燥棒状杆菌分别是tet(X)和poxtA基因的潜在宿主,证明环境暴露后猪场工人皮肤丰度显著升高的ARGs是通过宿主微生物携带,在其获得AMR后在工人皮肤和环境之间传播。结合皮肤微生物群和耐药基因组变化分析结果,可以得出结论,从环境暴露5小时开始,猪场环境中携带ARGs的宿主微生物可以稳定停留在工人皮肤,至其工作结束返回居住地。目前猪场硬性要求工人进场前需全身清洗消毒,但出场时无要求,因此猪场环境中的ARGs存在由工人携带向周围环境和社区传播的潜在风险。这提示猪场需要改进生产工作模式,比如可以在出口设置淋浴消毒房;由于灰尘对面部皮肤微生物的影响,还需要按时清洁通风口的灰尘,从而减少ARGs和ARB的传播。
结 论
本研究表明家畜环境中的职业暴露动态地重塑了人的皮肤微生物组和耐药组。这种重塑可在数小时内发生,并且获得的病原体和耐药基因有可能以农场工人作为载体传播到一般人群。在同一个健康框架下,应特别考虑农场工人的作用,以遏制抗微生物药物耐药性的蔓延。
代码和数据可用性
支持本文结论的宏基因组测序数据已存入NCBI Sequence Read Archive,项目ID为PRJNA982563(https://www.ncbi.nlm.nih.gov/bioproject/PRJNA982563/)。本文使用的数据和脚本保存在GitHub https://github.com/cdr67/Daily-occupational-exposure-in-swine-farm-alters-human-skin-microbiota-and-antibiotic-resistome中。补充材料(图表、表格、脚本、图形摘要、幻灯片、视频、中文翻译版本和更新材料)也可从线上获取。
。
代码和数据可用性
Dong-Rui Chen, Ke Cheng, Lei Wan, Chao-Yue Cui, Gong Li, Dong-Hao Zhao, Yang Yu, Xiao-Ping Liao, Ya-Hong Liu, Alaric W. D'Souza, Xin-Lei Lian, Jian Sun. 2024. Daily occupational exposure in swine farm alters human skin microbiota and antibiotic resistome. iMeta 3, e158. https://doi.org/10.1002/imt2.158
作者简介

陈东瑞(第一作者)
● 华南农业大学基础兽医学硕士,导师刘雅红教授,现就职于广西农垦永新畜牧集团牧业科技有限公司。
● 主要研究方向为微生物组学,相关学术成果已发表于iMeta、Frontiers in Microbiology等期刊。

程珂(第一作者)
● 博士研究生学历,博士后,兽医师,中国兽医协会诊断分会副会长,本硕博均就读于华南农业大学,导师刘雅红教授,现任广西农垦永新畜牧集团有限公司生产技术部副部长、牧业研究院副总经理、兽医总监。
● 主要从事于广西农垦永新畜牧集团猪场生产技术管理、猪群健康管理和猪场生物安全管理等工作,曾参与十三五国家重点研发项目、国家自然科学基金重点项目等。先后在Nature Communications、Frontiers in Microbiology、Vet Microbiol等国际期刊发表SCI论文8篇。

孙坚(通讯作者)
● 博士,华南农业大学兽医学院博士生导师,《中国兽医杂志》、One Health Advances等杂志编委。
● 主要研究动物源细菌耐药性产生、传播机制和防控技术。主持国家自然科学基金等省部级课题10项,近五年以第一或通讯作者在Nature Microbiology、Nature Communications、Science Advances、iMeta等国际刊物上共发表研究论文43篇。

连新磊(通讯作者)
● 华南农业大学副研究员,硕士生导师。
● 主要研究方向为生物信息学、基因转录调控与细菌耐药性,以第一作者或通讯作者(含共同)在Nucleic Acids Research、PLoS Genetics、Science of the Total Environment等期刊发表学术论文8篇,主持国家自然科学基金青年基金项目、广东省自然科学基金面上项目及广州市基础研究计划基础与应用基础研究项目等,相关成果申请2项发明专利。

Alaric W. D’Souza(通讯作者)
● 波士顿儿童医院儿科住院医师,圣路易斯华盛顿大学医学院医学博士。
● 主要研究细菌生态学、抗微生物药物耐药性和基因组学。近五年以第一或通讯作者在Clinical Infectious Diseases、Nature Communications、Genome Medicine等国际刊物上共发表研究论文6篇。
更多推荐
(▼ 点击跳转)
iMeta | 引用7000+,海普洛斯陈实富发布新版fastp,更快更好地处理FASTQ数据

iMeta | 德国国家肿瘤中心顾祖光发表复杂热图(ComplexHeatmap)可视化方法

1卷1期

1卷2期

1卷3期

1卷4期

2卷1期

2卷2期

2卷3期

2卷4期
期刊简介
“iMeta” 是由威立、肠菌分会和本领域数百位华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表原创研究、方法和综述以促进宏基因组学、微生物组和生物信息学发展。目标是发表前10%(IF > 15)的高影响力论文。期刊特色包括视频投稿、可重复分析、图片打磨、青年编委、50万用户的社交媒体宣传等。2022年2月正式创刊发行!目前期刊已经被ESCI、Scopus等数据库收录。
联系我们
iMeta主页:http://www.imeta.science
出版社:https://onlinelibrary.wiley.com/journal/2770596x
投稿:https://mc.manuscriptcentral.com/imeta
邮箱:office@imeta.science


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









