




基于GraftM对功能基因进行物种注释
Taxonomic classification of microbes with a given function based on a specific functional gene
赵圣国1, *
1动物营养学国家重点实验,中国农业科学院北京畜牧兽医研究所院,北京
*通讯作者邮箱:zhaoshengguo@caas.cn
引用格式:赵圣国. (2021). 基于GraftM对功能基因进行物种注释. // 微生物组实验手册. Bio-101: e2104000. DOI: 10.21769/BioProtoc.2104000.
Yunyun Gao, Kai Peng, Defeng Bai, et al. 2024. The Microbiome Protocols eBook initiative: Building a bridge to microbiome research. iMeta 3: e182. https://doi.org/10.1002/imt2.182
摘要:功能微生物是指执行某一特定功能的一类微生物群体。与一般性微生物相比,功能微生物与生态位表型具有更直接的联系,更能反映出生态位的功能变化。因此研究功能微生物多样性,对于解析生态位的功能机制具有重要意义。常用的RDP Classifier等算法无法适用于功能基因物种注释分析,因此本文介绍了基于GraftM的系统发育树原理对功能基因进行物种注释的方法。
关键词: GraftM,功能微生物,功能基因,物种注释
研究背景:
微生物多样性分析中,物种注释是最为关键的步骤。对于微生物多样性分析,常使用16S rRNA基因或ITS序列,利用RDP Classifier[1]等通过朴素贝叶斯算法对序列进行物种注释。功能微生物是指执行某一特定功能的一类微生物群体,比如产甲烷微生物、尿素分解微生物、氨氧化微生物、固氮微生物。与一般性微生物相比,功能微生物与生态位表型具有更直接的联系,更能反映出生态位的功能变化。因此研究功能微生物多样性,对于解析生态位的功能机制具有重要意义。功能微生物多样性研究中,常对某些关键功能基因进行测序分析。与16S rRNA基因或ITS基因相比,功能基因常具有多个不同拷贝,难以作为系统发育的标签基因,无法根据基因序列组成和相似特点直接进行物种注释,所以常用的RDP Classifier等算法无法适用于功能基因物种注释分析。GraftM[2]是用于功能基因注释的优秀软件,它通过对已知功能基因构建系统发育树 (含物种信息),然后将查询功能基因定位到系统发育树,根据树上位置和距离,注释查询功能基因物种信息。本文介绍了基于GraftM进行功能微生物的物种注释。
软件和数据库
Graftm (0.13.1) ( https://pypi.org/project/graftm/)
Bioconda ( https://bioconda.github.io/)
实验步骤
一、安装Graftm程序
通过conda安装:
conda create -n graftm
conda activate graftm
conda install graftm -c bioconda
二、创建与更新功能基因数据库包
1.下载功能基因数据
登录NCBI核酸数据库 (https://www.ncbi.nlm.nih.gov/nuccore),根据功能基因 名称查询序列,下载目标功能基因序列和物种分类信息,分别整理成两个文件 (m arker.genes.fasta和marker.genes.taxonomy.txt)(图1和图2)。
文件1:参考功能基因文件,marker.genes.fasta,格式为FASTA:

图1. 参考功能基因文件格式
文件2:参考功能基因物种信息文件,marker.genes.taxonomy.txt,文本文件(第一列为ID,第二列为分类信息,两列Tab隔开),格式如下:

图2. 参考功能基因物种信息文件格式
例子:以搜索脲酶基因ureC为例[3]
1)登录NCBI核酸数据库,输入关键词“ureC”,检索后出现所有包含ureC基因的序列或基因组。点击需要下载的序列,进入信息页(图3)。
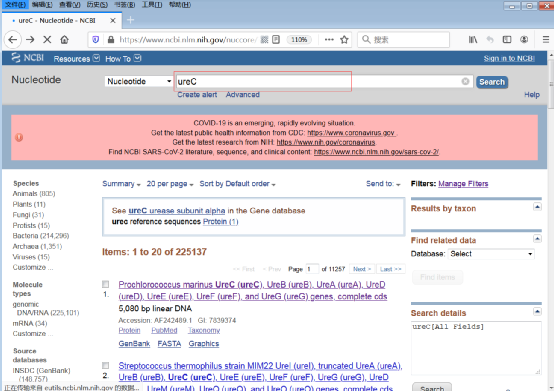
图3:NCBI核酸数据库,需要下载序列信息页
2)找到ureC基因所在的编码位置,本例中是1 – 1710(图4)。
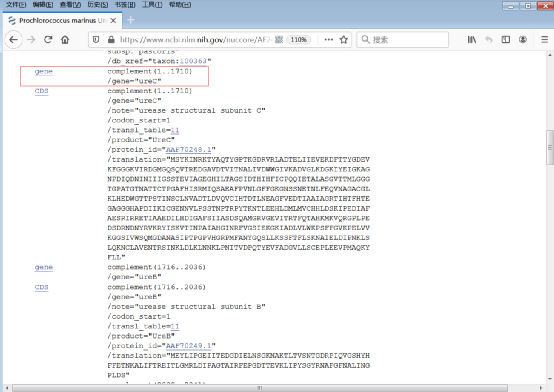
图4:ureC基因所在的编码位置
3)鼠标滑轮上滑后,在“Change region shown”那里输入1 - 1710,点击update view(图5)。
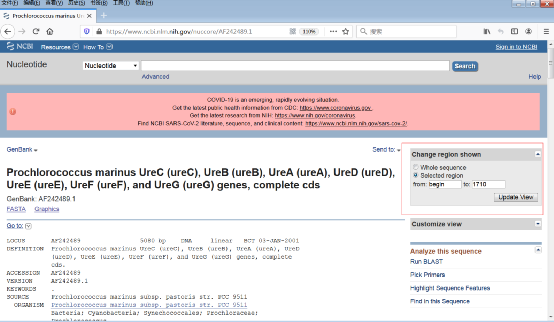
图5:“Change region shown”界面
4)保存ORGANISM信息(图6)。
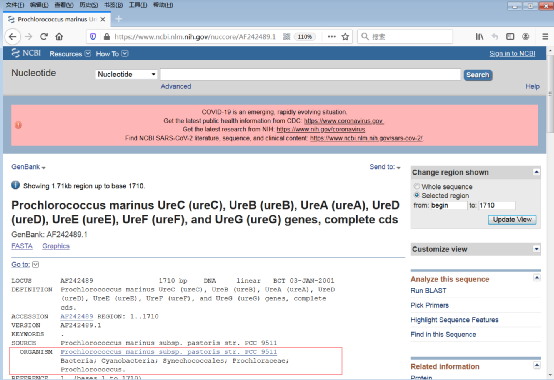
图6:ORGANISM信息界面
5)点击显示方式为FASTA,将FASTA格式序列保存(图7)。
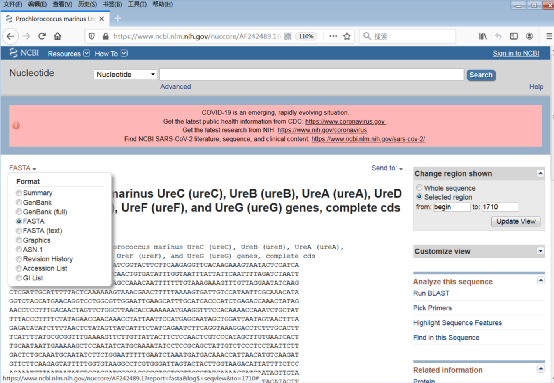
图7:保存FASTA格式
6)将所有下载的ureC基因FASTA序列复制到一个文件中,物种分类信息复制到另一个文件中。
两个文件格式为(图8,9):
文件1:参考功能基因文件,格式为FASTA:
 图8:参考功能基因文件
图8:参考功能基因文件
文件2:参考功能基因物种信息文件,文本文件(第一列为ID,第二列为分类信息,两列Tab隔开):

图9:参考功能基因物种信息文件
2.创建功能基因数据库包
运行程序:
graftM create --sequences marker.genes.fasta --taxonomy marker.genes.taxonomy.txt --output marker.genes.gpkg(图10):
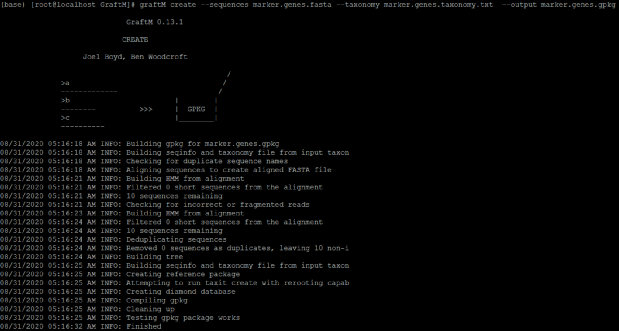
图10. 运行结果
graftM create参数:
--sequences;参考功能基因序列文件,必选
--taxonomy;参考功能基因物种信息文件,必选
--alignment;比对后文件,如果有可提交,以减少运行时间
--hmm;HMM文件,如果有可提交,以减少运行时间
--tree;newick格式的系统发育树文件,同时提供log文件
--tree_log;系统发育树的log文件
--output;输出文件夹
--threads;线程数
--graftm_package;需要更新的旧数据库包,仅更新数据库包时使用
3.更新数据库包
如果新下载功能基因需要补充到数据库中,则需要更新数据库包。
运行程序:
graftM create --graftm_package marker.genes.gpkg --sequences marker.genes.new.fasta --taxonomy marker.genes.new.taxonomy.txt --output marker.genes.updated.gpkg
三、功能基因物种注释
运行程序:
graftM graft --forward query.fasta --graftm_package marker.genes.gpkg/ --output_directory query.graftm
graftM graft参数:
--forward;查询功能基因序列,fasta格式,必选
--graftm_package;构建好的数据库包,必选
--output;输出文件夹
--threads;线程数 (默认5)
--placements_cutoff confidence;置信截取值 (默认0.75)
结果与分析
导出文件夹query.graftm中query文件夹中query_read_tax.tsv文件。第一列为OTU (Feature) 编号,第二列为分类信息,如下所示(图11):

图11. 运行结果
致谢
感谢中国农业科学院创新工程 (ASTIP-IAS12) 支持。
参考文献
[1] Wang, Q, G. M. Garrity, J. M. Tiedje, and J. R. Cole. (2007). Naïve Bayesian Classifier for Rapid Assignment of rRNA Sequences into the New Bacterial Taxonomy. Appl Environ Microbiol. 73: 5261-5267.
[2] Joel A Boyd, Ben J Woodcroft and Gene W Tyson. (2018). GraftM: a tool for scalable, phylogenetically informed classification of genes within metagenomes. Nucleic Acids Research. 46(10): e59.
[3] Jin, D., Zhao, S., Zheng, N., Bu, D., Beckers, Y., Denman, S. E., McSweeney, C. S. and Wang, J. (2017). Differences in ureolytic bacterial composition between the rumen digesta and rumen wall based on urec gene classification. Front Microbiol 8: 385.
宏基因组推荐
本公众号现全面开放投稿,希望文章作者讲出自己的科研故事,分享论文的精华与亮点。投稿请联系小编(微信号:yongxinliu 或 meta-genomics)
猜你喜欢
iMeta高引文章 fastp 复杂热图 ggtree 绘图imageGP 网络iNAP
iMeta网页工具 代谢组MetOrigin 美吉云乳酸化预测DeepKla
iMeta综述 肠菌菌群 植物菌群 口腔菌群 蛋白质结构预测
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature
一文读懂:宏基因组 寄生虫益处 进化树 必备技能:提问 搜索 Endnote
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流快速解决科研困难,我们建立了“宏基因组”讨论群,己有国内外6000+ 科研人员加入。请添加主编微信meta-genomics带你入群,务必备注“姓名-单位-研究方向-职称/年级”。高级职称请注明身份,另有海内外微生物PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。
点击阅读原文

















 1138
1138

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









