







土壤线虫群落DNA提取、扩增及高通量测序
DNA extraction, amplification and high-throughput sequencing of soil nematode community
杜晓芳1,2,梁文举1,李琪1*
1中国科学院沈阳应用生态研究所,土壤生态与农业生态工程研究中心,沈阳,110016
2中国科学院大学,北京,100049
*通讯作者邮箱:liq@iae.ac.cn
引用格式:杜晓芳, 梁文举, 李琪. (2021). 土壤线虫群落DNA提取、扩增及高通量测序. // 微生物组实验手册. Bio-101: e2104085. DOI: 10.21769/BioProtoc.2104085.
How to cite: Yunyun Gao, Kai Peng, Defeng Bai, et al. 2024. The Microbiome Protocols eBook initiative: Building a bridge to microbiome research. iMeta 3: e182. https://doi.org/10.1002/imt2.182
摘要:应用高通量测序技术开展土壤线虫多样性研究,能够克服形态学鉴定所需的时间长和专业知识的限制,为在大尺度上开展土壤线虫生态学研究提供了技术保障。虽然高通量测序技术可以快速、高效、较为准确的分析大量土壤样品的线虫群落组成,但利用土壤试剂盒提取线虫DNA并进行高通量测序得到的测序深度较低,线虫序列占真核生物总序列的比例较小。本文针对目前利用高通量测序技术开展线虫研究存在的线虫富集以及线虫DNA提取等方面的问题,对提取土壤线虫DNA的方法进行了改进。通过对线虫提取方法的优化和线虫试剂盒的筛选,研究结果表明:浅盘法结合贝尔曼漏斗法对线虫进行提取、物理去杂,并使用DNeasy Blood & Tissue试剂盒提取线虫DNA,能够提高线虫的提取效率和测序深度,这为利用高通量测序技术开展土壤线虫群落研究提供了技术参考。
关键词:土壤线虫,DNA提取,高通量测序
材料与试剂
1.一次性口罩、无菌手套
2.无水乙醇
3.0.5 ml和2 ml的离心管
4.10 μl、20 ~ 200 μl、1000 μl的移液器(Eppendorf)
5.DNeasy Blood & Tissue 线虫DNA提取试剂盒
6.NF1/18Sr2b引物
7.线虫提取富集相关材料(18目、60目、500目不锈钢网筛(浙江上虞银河测试仪器厂)、面巾纸、表面皿、天平、烧杯、2 L的量杯等)
仪器设备
1.4/-80 ℃冰箱(海尔)
2.真空泵(郑州长城科工贸有限公司SHB-lll)
3.高压灭菌锅(上海申安医疗器械厂DSX-18L-l)
4.恒温水浴锅(上海精宏实验设备有限公司DK-S24)
5.无菌工作台
6.离心机(Eppendorf Centrifuge 5425)
7.PCR仪(ABI GeneAmp® 9700型)
实验步骤
一、土壤线虫的提取富集
线虫的提取富集使用浅盘法结合贝尔曼漏斗的方法进行(图1)。
1.取100 g鲜土倒入量杯中,每次1 L水淘洗,悬停1分钟,倒入上层60目下层500目的一组网筛,重复三次。
2.将淘洗后500目网筛上得到的泥浆转移至有面巾纸的18目网筛中(网筛置于浅盘),静置24 h。
3.静置24 h后取走网筛,将浅盘中的水全部转移至250 ml的烧杯中。将烧杯中的线虫悬液经玻璃棒引流转移至铺有面巾纸的漏斗中(直径10 ~ 15 cm),静置6 h以上。
4.打开漏斗胶管下部的弹簧夹收集线虫悬液,静置2 h后用真空泵抽取上清液,留约5 - 10 ml的线虫悬液在显微镜下活体计数。线虫悬液可置于4 ℃冰箱保存一周(或室温保存1-2天)。
二、土壤线虫DNA提取
1.在提取线虫DNA之前,将每个样品的线虫悬液以1902 rcf/min离心10分钟(或线虫悬液静置2h以上)。弃去上清液后,保留约2 ml线虫悬浮液并转移到2 ml离心管中。然后将2 ml离心管以6010 rcf/min离心2分钟,弃去上清液后,最终保留0.5 ml线虫悬液使用DNeasy Blood & Tissue Kit试剂盒(Qiagen)进行线虫DNA提取。
2.根据DNeasy Blood & Tissue Kit试剂盒的说明书,我们对实验方法进行了微调,为了得到更多的线虫DNA,我们使用双倍的裂解缓冲液(360 μl Buffer ATL, 40 μl蛋白酶K)来充分浓缩和裂解线虫,消化时间延长至1.5 h。具体的操作步骤如下:
j 在保留0.5 ml线虫悬液的离心管中加入360 μl Buffer ATL,40 μl蛋白酶K,涡旋15 s后水浴消化1.5h,水浴消化期间间断振荡(每隔15min上下颠倒几次);
k 涡旋15 s,加入400 μl Buffer AL,涡旋混匀后加入400 μl(96%-100%)的酒精,涡旋混匀;
l 用移液器将2 ml离心管中的所有液体转移至带有滤柱的2 ml收集管中(分几次转移视情况而定),6010 rcf离心1 min,弃去离心液及收集管;
m 将滤柱转移到一个新的收集管中,加入500 μl Buffer AW1,6010 rcf离心1 min,弃去滤出液和收集管;
n 将滤柱转移到一个新的收集管中,加入500 μl BufferAW2,18407 rcf离心3 min干燥滤柱,弃去滤出液和收集管;
o 将滤柱置于一个1.5 ml的离心管中,加入100 μl(一般为50 μl-200 μl)Buffer AE,室温孵化1 min后6010 rcf离心1min最终得到线虫DNA;
p 提取的线虫DNA储存在-80 ℃冰箱用于后续的PCR扩增和测序。
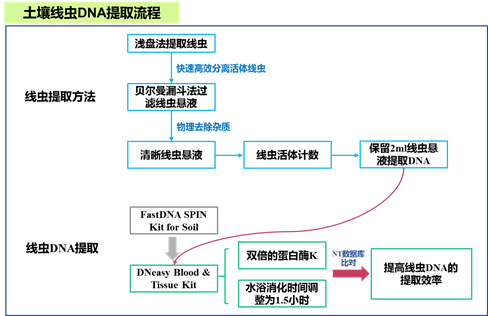
图1. 土壤线虫DNA提取流程示意图。(相关结果来自课题组先前发表的文章(Du et al.,2020))
三、线虫群落的扩增测序
1.使用引物NF1-F/18Sr2b-R(Porazinska et al., 2009)对线虫18S rDNA V4区进行扩增。PCR采用TransGen AP221-02: TransStart Fastpfu DNA Polymerase,20 μl反应体系:5 × FastPfu Buffer 4 μl,2.5 mM dNTPs 2 μl,Forward Primer (5μM)0.8 μl,Reverse Primer (5μM)0.8 μl,FastPfu Polymerase 0.4 μl,BSA 0.2 μl,Template DNA 10 ng,最后使用灭菌PCR水补足至20 μl。
2.线虫(NF1FF/18Sr2bR)PCR反应参数为:95 ℃预变性3 min,95 ℃变性30 s,55 ℃退火30 s,72 ℃延伸45 s,35次循环后最终在72 ℃终延伸10 min。扩增之后,PCR产物使用2 %琼脂糖凝胶电泳进行可视化(图2),使用AxyPrep DNA凝胶提取试剂盒(Axygen Biosciences, Union City, CA, USA)进行纯化,并使用QuantiFluor™-ST(Promega, USA)对回收产物进行检测定量。根据Illumina MiSeq平台(Illumina, San Diego, USA)标准操作规程将纯化后的扩增片段构建PE 300文库。PCR文库构建是在引物上合成barcode,采用混合样品建库的模式。具体构建文库的步骤为: (1)连接“Y”字形接头;(2)使用磁珠筛选去除接头自连片段;(3)利用PCR扩增进行文库模板的富集;(4)氢氧化钠变性,产生单链DNA片段。利用Illumina公司的Miseq PE300平台进行双端测序。原始数据已上传至NCBI数据库(序列号:PRJNA580055)。
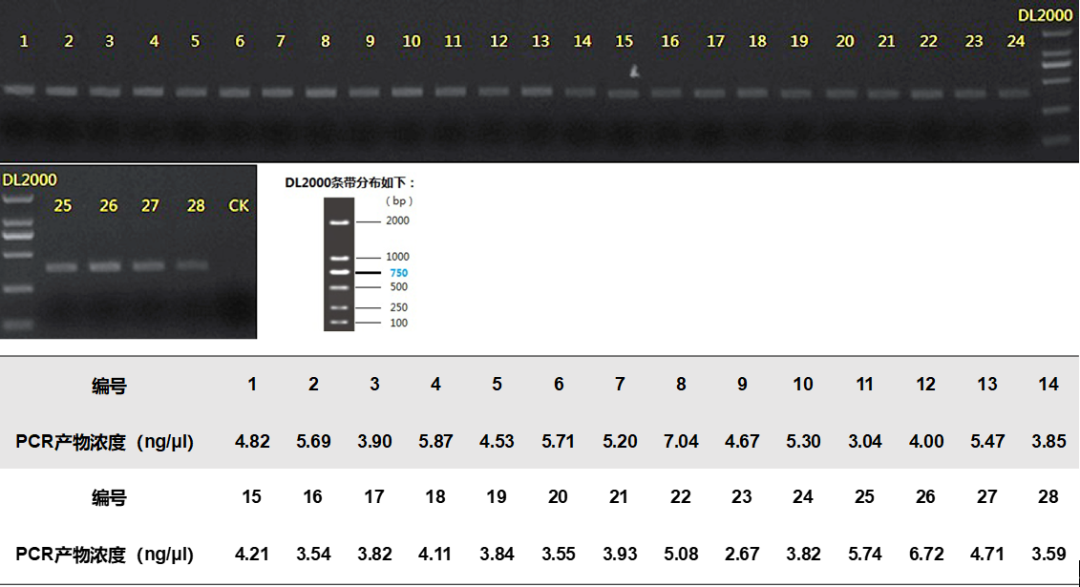
图2. 利用NF1和18Sr2b引物对进行PCR扩增的电泳图谱及PCR产物浓度。(相关结果来自课题组先前发表的文章(Du et al.,2020))
结果与分析
一、生物信息分析
1.数据优化
使用Trimmomatic软件对原始测序序列进行质控,使用FLASH(http://www.cbcb.umd.edu/software/flash,version 1.2.7)软件进行拼接:
(1)过滤reads尾部质量值20以下的碱基及质控后200bp以下的reads,去除含N碱基的reads;
(2)根据PE reads之间的overlap关系,将成对reads拼接成一条序列,最小overlap长度为10bp;
(3)拼接序列的overlap区允许的最大错配比率为0.2,去除无法拼接的序列;
(4)根据序列首尾两端的barcode和引物区分样品,并调整序列方向,barcode需精确匹配,引物允许2个碱基的错配。
2. OTU聚类
使用UPARSE软件(http://drive5.com/uparse/,version 7.1),根据 97%的相似度对序列进行OTU聚类,具体流程如下:
(1)提取优化序列中的非重复序列,去除没有重复的单序列;
(2)按照97%相似性对非重复序列(不含单序列)进行OTU聚类,在聚类过程中去除嵌合体,得到OTU代表序列;
(3)将所有优化序列map至OTU代表序列,选出与OTU代表序列相似性在97%以上的序列,生成OTU表格。
3. 分类学分析
通过Blast搜索,比对NCBI NT数据库对物种进行分类注释。
二、线虫DNA提取效率和测序深度比较
利用浅盘法结合贝尔曼漏斗法提取富集线虫后,利用DNeasy Blood & Tissue试剂盒提取线虫DNA,注释的线虫序列占总序列的68.40%,平均每个样本的测序深度达到28978/样本。相比前人的研究结果(Sapkota et al., 2015; Peham et al., 2017; Griffiths et al., 2018; Treonis et al., 2018)(表1),经过对线虫的物理分离、富集以及试剂盒提取方法的改进,提高了线虫DNA的提取效率和测序深度,为利用高通量测序技术开展土壤线虫群落研究提供了技术参考。
表1. 基于高通量测序不同提取方法线虫测序深度比较
线虫DNA提取试剂盒 | 测序深度 (序列数) | 线虫占 真核生物比例 | 参考文献 |
the PowerLyzer™Power Soil® DNA Isolation Kit | 3994/样本 | 64.40% | Sapkota and Nicolaisen, 2015 |
PowerSoil® DNA Isolation Kit | 未提到 | 2.50% | Peham et al., 2017 |
PowerMax Soil DNA isolation kit | 2175/样本 | 未提到 | Griffiths et al., 2018 |
PowerSoil® DNA Isolation Kit | 2175/样本 | ||
MO BIO UltraClean® Tissue & Cells DNA Isolation kit | 32568/样本 | 19.9% | Treonis et al., 2018 |
DNeasy Blood & Tissue Kit | 28978/样本 | 68.40% | Du et al., 2020 |
注意事项
1. 针对不同的土壤选择合适的土壤线虫提取方法是提取线虫DNA的关键步骤,在保证线虫富集效率的同时还要保证线虫悬液样本的清晰,土壤线虫提取方法的选择可参考张晓珂 等(2013)。
2. 线虫引物和试剂盒在未来可能会进一步优化,需关注最新的研究进展选择合适的引物和试剂盒进行研究。
3. 线虫高通量测序数据的处理可根据需求灵活分析,也可使用Usearch软件对原始测序序列进行质控,聚类,生成OTU表。
4. 用于线虫分析的数据库对于分子技术在线虫领域的应用十分重要,今后需进一步补充和完善现有的线虫数据库。
致谢
本研究成果主要来源于课题组先前发表的相关文章(Du et al.,2020)。相关研究得到了国家科技基础资源调查专项项目(2018FY100304)、中国科学院国际合作局对外合作重点项目(151221KYSB20200014)和国家自然科学基金项目(41877047)的资助。
参考文献
1.张晓珂, 梁文举, 李琪. (2013) 长白山森林土壤线虫. 北京:中国农业出版社.
2.Du, X. F., Li, Y. B., Han, X., Ahmad, W. and Li, Q. (2020). Using high-throughput sequencing quantitatively to investigate soil nematode community composition in a steppe-forest ecotone. Appl Soil Ecol. 152: 103562.
3.Griffiths, B. S., de Groot, G. A., Laros, I., Stone, D. and Geisen, S. (2018). The need for standardisation: exemplified by a description of the diversity, community structure and ecological indices of soil nematodes. Ecol. Indic. 87: 43-46.
4.Peham, T., Steiner, F. M., Schlick-Steiner, B. C., and Arthofer, W. (2017). Are we ready to detect nematode diversity by next generation sequencing?. Ecol Evol. 7: 4147-4151.
5.Porazinska, D. L., Giblin-Davis, R. M., Faller, L., Farmerie, W., Kanzaki, N., Morris, K., Powers, T. O., Tucker, A. E., Sung, W., and Thomas, W. K. (2009). Evaluating high-throughput sequencing as a method for metagenomic analysis of nematode diversity. Mol Ecol Resour. 9: 1439-1450.
6.Sapkota, R. and Nicolaisen, M., (2015). High-throughput sequencing of nematode communities from total soil DNA extractions. Bmc Ecol 15: 3.
7.Treonis, A. M., Unangst, S. K., Kepler, R. M., Buyer, J. S., Cavigelli, M. A., Mirsky, S. B. and Maul, J. E. (2018). Characterization of soil nematode communities in three cropping systems through morphological and DNA metabarcoding approaches. Sci Rep-Uk. 8: 2004.
宏基因组推荐
本公众号现全面开放投稿,希望文章作者讲出自己的科研故事,分享论文的精华与亮点。投稿请联系小编(微信号:yongxinliu 或 meta-genomics)
猜你喜欢
iMeta高引文章 fastp 复杂热图 ggtree 绘图imageGP 网络iNAP
iMeta网页工具 代谢组MetOrigin 美吉云乳酸化预测DeepKla
iMeta综述 肠菌菌群 植物菌群 口腔菌群 蛋白质结构预测
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature
一文读懂:宏基因组 寄生虫益处 进化树 必备技能:提问 搜索 Endnote
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流快速解决科研困难,我们建立了“宏基因组”讨论群,己有国内外6000+ 科研人员加入。请添加主编微信meta-genomics带你入群,务必备注“姓名-单位-研究方向-职称/年级”。高级职称请注明身份,另有海内外微生物PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。
点击阅读原文

















 974
974

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









